INTRODUCTION
Interferons (IFNs) are pleiotropic cytokines with antiviral, antitumor and immunomodulatory properties, being central coordinators of the immune response (1). The term “interferons” comes from the description of molecules protecting cells by “interfering” with viral infection (2, 3).
Three major types of IFNs are distinguished by their sequence identity, genetic loci, cell of origin, nature, and distribution of their receptors and resulting stimuli (Table 1).
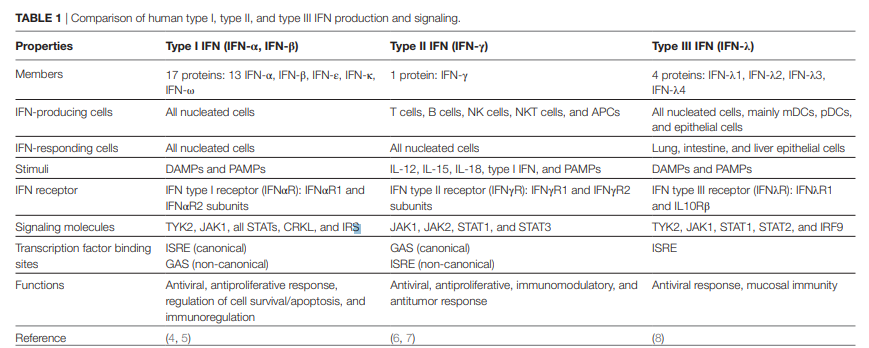
The human type I IFN family comprises 17 distinct proteins, mainly represented by IFN-α and IFN-β, which are ubiquitously expressed and signal through their cognate receptor, composed by IFNαR1 and IFNαR2 subunits [reviewed in Ref. (4)].
IFN-γ is the lone member of type II IFN family. It is more restrictively expressed and is structurally and functionally different from the other types of IFNs. Most recently, a type III IFN family was described to be composed of four homologous proteins (IFNλ1–4), which bind the IFNλR1 and interleukin (IL)-10Rβ heterodimeric receptor [reviewed in Ref. (8)].
To date, type I and type III IFNs have been mainly involved in host–pathogen interactions, and their expression is activated through immune system sentinel receptors, such as pattern recognition receptors. Despite the similar function of type I and III on antiviral infections, it is the viral tropism that dictates the relative contribution of each IFN (9).
Moreover, whereas almost all nucleated cells respond to type I IFN, type III IFNs response is restricted to tissues with a high risk of viral exposure and infection, as the mucosal surfaces. The role of type II IFN in promoting host immune response to microorganisms is similarly well documented. Notably, it is also known to play a pivotal function on cancer immune surveillance, stimulating antitumor immunity and promoting tumor recognition and elimination (10–16).
This review focuses on type II IFN signaling, cellular functions, and directed therapies and was encouraged by novel findings revealing regulatory mechanisms of IFN-γ and its prognostic as well as therapeutic potential. In fact, since Wheelock who reported that IFN-γ inhibited viral replication in 1965 (17), it took around 30 years to envisage this cytokine as a target of antitumor immunity (18).
Interferon-gamma is a homodimer formed by the noncovalent association of two 17 kDa polypeptide subunits. During synthesis, after multiple N-glycosylation, both subunits bind in an antiparallel manner, constituting a mature 50 kDa molecule (19, 20).
Notably, the IFN-γ symmetry suggests that a single molecule can bind simultaneously to two receptors, amplifying the underlying responses. Cellular responses induced by IFN-γ may also involve cross-communication with IFN-α/β receptors, amplifying IFN-γ signaling and its effects (21, 22). Interferon-gamma is secreted predominantly by activated lymphocytes such as CD4 T helper type 1 (Th1) cells and CD8 cytotoxic T cells (23–26), γδ T cells (27–33), and natural killer (NK) cells (34, 35) and, to a less extent, by natural killer T cells (NKT), B cells (36–39), and professional antigen-presenting cells (APCs) (40–42). Its expression is induced by mitogens and cytokines, such as IL-12 (43, 44), IL-15 (45), IL-18 (46, 47), and type I IFN (48, 49).
IFN-γ pleiotropic functions are mediated by cell-specific expression of hundreds of IFN-γ-regulated genes that encompass inflammatory signaling molecules, apoptosis and cell cycle regulators, and transcriptional activators (50). Autocrine IFN-γ produced by APCs can act locally and contribute to sustain self and neighbor cell activation (51–53), crucial for early control of pathogen spreading, while T lymphocytes are the major paracrine source of IFN-γ in adaptive immunity. Under physiological conditions, the constitutive expression of type I and II IFNs is tightly controlled, remaining localized to tissues, without systemic effects (54–56). For instance, constitutive expression of endogenous IFN-γ contributes to the homeostasis of immune cell functions (57), maintenance of the hematopoietic stem cell niche (58), and bone formation (59). Combination approaches to boost innate immune activation have been explored to converge onto IFN pathways. However, IFN-γ-related signaling can also have suppressive immunoregulatory effects on antiviral (60, 61), autoimmune (62, 63), as well as on antitumor responses (64, 65). Unveiling cellular targets of IFN-γ is critically important for its therapeutic application, to predict patient responses, particularly in cancers where this cytokine can exert protumorigenic effects. Therefore, the cellular and molecular effects of IFN-γ, with particular emphasis on its dual role on tumor immunity and how to overcome its limitations, will be the major focus of this review.
Canonical signaling and regulatory mechanisms
The IFN-γ Receptor
The IFN-γ receptor is composed of two ligand-binding IFNγR1 chains associated with two signal-transducing IFNγR2 chains, which are responsible for connecting to the cytoplasmic transduction machinery (see Figure 1).
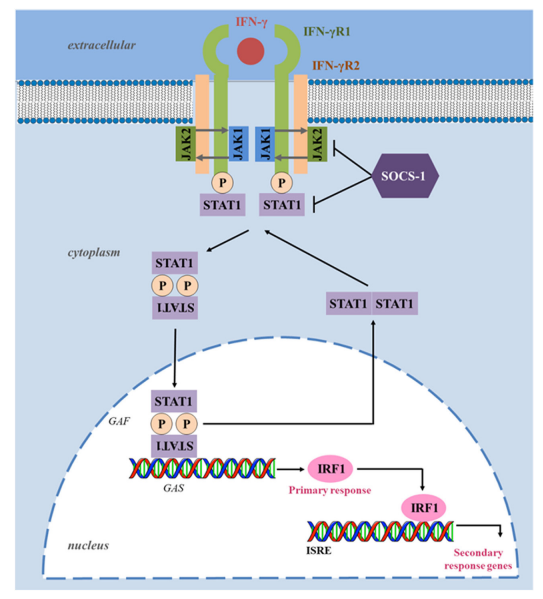
FIGURE 1 | Interferon-gamma (IFN-γ) canonical signaling pathway. Upon ligand binding, IFNγR1 and IFNγR2 oligomerize and transphosphorylate, activating Janus activated kinase (JAK) 1 and JAK2. These, in turn, phosphorylate IFNγR1, creating a docking site for the signal transducer and activator of transcription (STAT) 1.
Phosphorylated STAT1 homodimerizes in an antiparallel configuration, forming a complex gamma-activated factor (GAF), which translocates to the nucleus and binds to gamma-activated site (GAS), located at the promoters of primary response genes, increasing their transcription. Upon induction, transcription factor interferonregulatory factor 1 (IRF1) binds to interferon-stimulated response element (ISRE) and enhances the transcription of several secondary response genes responsible for several immunomodulatory functions.
Suppressor of cytokine signaling (SOCS) proteins negatively regulate the IFN-γ pathway by inhibiting JAKs and STAT1 phosphorylation. Through dephosphorylation and deacetylation, the configuration of STAT1 homodimers reverts to parallel, triggering their exit from the nucleus.
The IFNGR1 and IFNGR2 are localized in chromosome 6 and 21, respectively, and their expression differs significantly. While IFNγR1 is constitutively expressed at moderate levels on the surface of almost all cells, IFNγR2 is constitutively expressed at low levels, and its expression is tightly regulated, according to the state of cellular differentiation or activation (66).
For example, CD4 T helper cell subsets differ in their ability to respond to IFN-γ (67, 68). Remarkably, IFN-γ activates the signal transducer and activator of transcription (STAT) 1 that maintains the expression of T-bet, the master transcription factor that controls IFN-γ expression in T cells (69).
This signaling constitutes a positive feedback loop that maximizes Th1 immunity (70–72). Notably, Th1 cells are more resistant to the antiproliferative effects of IFN-γ than Th2 cells. This is likely due to lower levels of expression of the IFNγR2 subunit that allows Th1 cells to continue to proliferate during IFN-γ signaling. By contrast, Th2 cells that do not produce IFN-γ express higher levels of the IFNγR2 subunit, rendering them particularly susceptible to the presence of IFN-γ that inhibits their proliferation (67, 68, 73). Nevertheless, IFNγR2 downregulation may be also induced in Th2 cells when they are exposed to IFN-γ (68).
Thus, IFN-γ appears to regulate the expression of its own receptor on specific cell types, representing a regulatory mechanism of cellular desensitization in response to cytokines present at the local microenvironment. As a result, IFNγR2 expression can be a limiting factor in IFN-γ responsiveness and functional outcome that can dictate the Th1–Th2 phenotype switch and modulate the subsequent immune response.
JAK/STAT Signaling Pathway
The biological effects of IFN-γ are elicited through activation of intracellular molecular signaling networks, mainly via the JAK/ STAT pathway, which modulates the transcription of hundreds of genes and mediates diverse biological responses (50, 74–76). Upon IFN-γ binding, the intracellular domains of IFNγR2 oligomerize and transphosphorylate with IFNγR1, activating the downstream signaling components, JAK1 and JAK2. The activated JAKs phosphorylate the intracellular domain of the receptor (tyrosine 440 on human IFNγR1), creating binding sites for STAT1 (77).
STAT1 is then phosphorylated in the C-terminus on tyrosine Y701 residues by JAK, resulting in the formation of STAT1 homodimers complexes, known as gamma-activated factors (GAFs), which translocate to the nucleus and regulate gene expression through binding to gamma-activated site (GAS) elements in the promoters of interferon-stimulated genes (ISGs) (78). One of the major primary response genes induced by STAT1 signaling is the transcription factor interferon-regulatory factor 1 (IRF1), a member of the IFN regulatory transcription factor family (79). IRF1 functions as a transcription activator of interferonstimulated response elements (ISRE), leading to the transcription of a large number of secondary response genes (Figure 1). For instance in breast cancer cells, a genome-wide identification of IFN-γ-induced IRF1 activation reveals over 17,000 binding sites, with “apoptosis” or “cell death” as the most enriched target processes underlying the direct tumoricidal property of the cytokine (80).
However, tumor cells also develop resistance to IFN-γ through differential IRF1 responsiveness, pointing out that the JAK/STAT signaling pathway needs to be tightly regulated to avoid detrimental consequences of excessive stimulation and highlighting its role on immune responses and tumorigenesis (81). STAT1 targets of the IFN-γ-mediated signaling also include the SMAD family member 7 (SMAD7), and proteins involved in cell cycle regulation, such as c-Myc and the cyclin-dependent kinase inhibitor 1A (82–84). The JAK/STAT signaling pathway is regulated at several levels by positive and negative mechanisms. In particular, deregulation or inhibition of the JAK/STAT pathway leads to lowered immunity and is often associated with increased tumorigenesis (85, 86) or metastatic dissemination (87).
STATs are also involved in the development and function of the immune system and play a role in maintaining tumor surveillance [reviewed in Ref. (88)]. STAT1, as a tumor suppressor, is deducted for its expression in tumor cells, modulates their immunological status and consequently their response to antitumor immune responses. Indeed, STAT1- deficient tumor cells were more susceptible to NK cells while STAT1-proficient tumor cells were more sensitive to CD8+ T cells (89). In the same way, STAT1-deficient mice that are impaired in Th1 cell polarization, exhibited reduced IFN-γ expression and compromised cytolytic and NK lytic activity, failing to control tumor growth in contrast with wild-type mice (90). In addition, cell-autonomous tumor-suppressor functions of STAT1 have also been reported in breast cancer (91). However, there is growing evidence that STAT1 also acts as a tumor promoter (92–94) since it can enhance resistance to chemotherapeutic agents and radiation in carcinoma (95).
Importantly, STAT1 also participates in the signaling from different cytokines, including IL-21, IL-27, and IL-35. These cytokines have been proposed to limit antitumor immunity in specific cellular, molecular, and microenvironmental contexts (96–101). Thus, STAT1 phosphorylation reflects not only the threshold and magnitude of IFN-γ response but also of other immune mediators, highlighting the importance of the regulation of STAT1 phosphorylation. One of the most important negative regulators of the JAK/STAT signaling pathway is the suppressor of cytokine signaling (SOCS) proteins, which expression is increased in response to IFN-γ signaling through IRF1 (102, 103). SOCS blocks the activity of JAKs by a negative feedback loop, but also regulates other cytokines downstream signaling. SH2 domains in SOCS proteins directly bind to phosphorylated tyrosine residues of activated JAKs, blocking the recruitment of signal transducer adaptors, such as STATs, and JAK activity (102). Furthermore, SOCS promote interactions that lead to ubiquitination and proteasome degradation of components of the JAK/STAT signaling (104, 105). SOCS1 even prevents regulatory T (Treg) cells from producing IFN-γ by suppression of STAT1, avoiding the conversion of Treg cells into effector cells (106). In addition, SOCS2-deficient mice showed a reduction in lung metastases and an increase in survival following melanoma challenge (107). Alternatively, the transcriptional activity of STAT1 can be positively regulated by other signaling cascades triggered by IFN-γ binding, such as the mitogen-activated protein kinase pathway, protein kinase C, and PI3K/AKT, which phosphorylate STAT1 in its transactivation domain (108). Adding to the complexity, under certain circumstances, IFN-γ also can activate STAT1-independent pathways through other transcription factors, namely STAT3 (109), STAT5 (110), nuclear factor-kappa B (NF-κB) (111), and activator protein 1 (112). In conclusion, the primary response of IFN-γ is mediated by GAF that acts on genes with GAS binding sequence in their promoter, while the primary response of type I IFNs is mediated by ISGF3 (STAT1/ STAT2/IRF9 complex) that induces genes that have ISRE in their promoter. Thus, some of the ISGs are regulated by both types of IFNs, whereas others are selectively regulated by each type of IFN, consequently potentiating the diversity of biological responses.
Biological functions
IFN-γ actions on immune Cells
Interferon-gamma signaling pathway coordinates several biological responses, primarily involved in host defense and immune surveillance but also in the establishment of adaptive immunity (Figure 2) and in the regulation of inflammation, apoptosis and cell cycle.

Figure 2 | Immunomodulatory effects of interferon-gamma (IFN-γ). IFN-γ produced by immune cells affects the behavior of distinct immune cells within the tumor microenvironment. Specifically, IFN-γ plays a major role in activating anticancer immunity, by promoting the activity of CD4 T helper type 1 cells, CD8 cytotoxic T lymphocyte (CTL), natural killer (NK) cells, dendritic cells (DCs), and macrophages, promoting the antigen presentation. Additionally, IFN-γ activates macrophages towards a more pro-inflammatory and tumoricidal phenotype (M1-like). Alternatively, IFN-γ inhibits regulatory T (Treg) cells, Th2 and Th17 differentiation and functions.
One of the first described biological effects of IFNs was the upregulation of the major histocompatibility complex (MHC) molecules (113, 114) as well as the upregulation of the whole MHC I and II antigen processing and presentation machinery including transporter associated with antigen processing (TAP) 1/2, invariant chain, and the expression and activity of the proteasome (115–122).
Furthermore, in some tumor types, such as multiple myeloma and melanoma cells, IFN-γ can also upregulate the MHC class II transactivator (CIITA) that leads to MHC class II expression (123, 124). Thus, IFN-γ initiates an immune-antigenic exposure program in the target cells, and this ensures the rapid recognition of stressed tissues. IFN-γ is a major product of Th1-mediated immune response and orchestrates Th1 effector mechanisms, as further activation of innate immunity (macrophages and NK cells) in a positive feedback loop. Upregulation of cell surface MHC class I by IFN-γ is crucial for host response to intracellular pathogens and tumor cells, due to cytotoxic T cell activation, promoting cell-mediated immunity. IFN-γ directly acts as a cytotoxic CD8 T cell differentiation signal, and it is essential for the induction of cytotoxic T cell precursor proliferation (125, 126).
IFN-γ also upregulates cell surface MHC class II on APCs, thus promoting peptide-specific activation of CD4 T cells (25, 127–129). In addition, IFN-γ activates macrophages toward a pro-inflammatory profile, exhibiting an increased phagocytic ability as well as enhanced microbial killing activity (130). In fact, IFN-γ was initially shown to induce “classical” activation of macrophages and polarization toward a tumoricidal phenotype (131). Interestingly, the original name of IFN-γ was macrophage activation factor (132, 133). IFN-γ controls specific gene expression programs involving more than 290 genes related to cytokine and chemokine receptors, cell activation markers, cellular adhesion proteins, MHC proteins, proteasome formation, protein turnover, and signaling mediators and regulators (134). The ability of IFN-γ to induce tumor cell killing includes the activation of the NADPH-dependent phagocyte oxidase system, nitric oxide production, tryptophan depletion and upregulation of lysosomal enzymes (121, 135, 136).
These events result in recruitment of effector cells to help in the inflammation resolution process (137, 138). In addition, as a major cytokine of Th1 cells, IFN-γ maintains Th1 lineage commitment through a positive feedback loop that stabilizes the Th cell phenotype (72, 139–141) and cross-inhibits the differentiation to other Th cell subsets (Figure 2). Indeed, IFN-γ inhibits Th2 cell differentiation (142, 143) and consequently IL-4 production. This regulation involves the inhibition of the IL-4/ STAT6 pathway, required for Th2 cell differentiation, and it is mediated at least by IFN-γ-induced SOCS1 that inhibits IL-4R signaling (144, 145). Furthermore, IFN-γ-induced T-bet inhibits Th2 cell differentiation by directly interfering with the activity of Th2 cell-specific transcription factor, GATA-3 (146). Höfer and colleagues, using mathematical models, proposed that IL-4 also acts to propagate Th2 cell differentiation (147). A high IL-4 level promotes increased GATA-3 expression that further enhances GATA-3 transcriptional imprinting for Th2 differentiation (147, 148). This model proposed that high expression state of GATA-3 can be suppressed by strong inhibition of autoactivation, as observed in the presence of Th1-polarizing conditions (147, 149). IFN-γ was also described to downregulate the IL-4-inducible gene expression (150). The cross-regulation of Th1 and Th2 cells was also demonstrated in STAT6-deficient mice, which lack Th2 phenotype and associated immune responses. These animals displayed augmented tumor-specific IFN-γ production and cytotoxic T cell activity and, consequently rejected the tumor cell line that grew progressively in the wild-type control (151). Interferon-gamma produced by Th1 cells also counteracts Th17 cell development and their effector functions (152–154). Several mechanisms can be considered as the inhibition of molecules involved in the Th17 differentiation (155, 156), the inhibition of STAT3 by STAT1 (157) and recently, T-bet was demonstrated to prevent differentiation of Th precursors into Th17 cells by blocking the expression of the Th17 cell lineagespecific transcription factor, RORγt (158). Furthermore, IFN-γ also exerts regulatory functions to limit tissue damage associated with inflammation (63, 159–162) (Figure 2). IFN-γ has been classically considered as a pro-inflammatory cytokine, involved in the regulation of anti-inflammatory responses, by antagonizing the IL-10 (157, 163) and TGF-beta (164) signaling pathways. Consequently, IFN-γ inhibits Treg cell differentiation and functions (165, 166). However, in some chronic inflammation conditions, IFN-γ plays a crucial role in attenuating tissue destruction. In this case, IFN-γ might be protective (62, 167) by promoting the number and function of Treg cells (168–170). In addition, IFN-γ production by Treg cells themselves was shown to be a key feature of the Treg cells that are capable of dampening Th1 cell responses (171–174). Thus, IFN-γ dictates the differentiation of specialized Foxp3+T-bet+ Treg cells that selectively suppress Th1 cells, and constitute a negative feedback loop to minimize the detrimental effect of IFN-γ. IFN-γ also promotes the differentiation of myeloid-derived suppressor cells (MDSCs) that restrain overactivation of effector T cells, maintaining tissue homeostasis (175, 176). Other regulatory mechanisms involving IFN-γ signaling that dampen the magnitude of the immune response have been reported, as the induction of indoleamine 2,3-dioxygenase (IDO) by Treg cells, monocytes and stromal cells (177–180), and of the programmed cell death 1 (PD-1) ligand (PD-L1) on immune and transformed cells, inhibiting T cell responses (181–183).
IFN-γ actions on Transformed Cells and on the Tumor Microenvironment
Interferon-gamma is involved in antiproliferative (18), antiangiogenic (184) and pro-apoptotic effects established against neoplastic cells. How IFN-γ induces the signaling pathways initiating and propagating the apoptotic cascade remains to be elucidated. The level of complexity is demonstrated by the fact that the mechanism might depend on the tumor cells themselves. For example, while in a glioblastoma cell line the induction of apoptosis was due to suppression of the PI3K/AKT pathway, in another glioblastoma cell line apoptosis occurred independently of the PI3K/AKT pathway but required NF-κB (185). It was also shown that IFN-γ induces apoptosis of human pancreatic carcinoma cells in a caspase-1-dependent manner (186). A review covered in detail the mechanism of induction of programmed cell death (187). So far, the known biological functions of IFN-γ indicate that, although it can act as a potent inducer of antitumor immunity, it actually has a dual role and may also favor tumor immune evasion.