Recombinant DNA Technology (With Diagram)
Recombinant DNA Technology (With Diagram)
ADVERTISEMENTS: In this article we will discuss about Recombinant DNA Technology:- 1.Steps in Recombinant DNA Technology 2. Tools for Recombinant DNA Technology 3. Techniques Used In Recombinant DNA Technology 4. Applications of Recombinant DNA Technology.
www.biologydiscussion.com
Techniques Used In Recombinant DNA Technology:
A number of techniques are used for various purposes during different steps of rec DNA technology.
Such techniques serve for the fulfilment of different requirements or to obtain proper information for drawing an exact inference during genetic engineering. Some of these important techniques are gel electrophoresis, blotting techniques, dot-blot hybridization, DNA sequencing, artificial gene synthesis, polymerase chain reaction, colony hybridization, etc.
fulfilment 수행
proper 적절한
exact 정확한
inference 추론
Gel Electrophoresis:
It is the technique of separation of charged molecules (in aqueous phase) under the influence of an electrical field so that they move on the gel towards the electrode of opposite charge i.e., cations move towards the negative electrode and anions move towards the positive electrode.
electrical field 전기적 영역
cation 양이온
anion 음이온
The genomic DNA is extracted from the desired host and is then fragmented using restriction endonucleases.
For separation of these cut fragments and isolation of desired DNA fragment, the technique of gel electrophoresis is employed. Gel electrophoresis may be of horizontal or vertical type.
extract 추출하다
fragment 조각
horizontal 수평의
vertical 수직의
Usually agarose gel is used for separation of large segments of DNA while the polyacrylamide gel is used for the separation of small DNA fragments which are only a few base pairs long.
segment 부분
Gel electrophoresis employs a buffer system, a medium which is a gel and a source of direct current (Fig. 6). Samples having DNA fragments are applied on the gel and current is passed through the system for an appropriate time. Different DNA fragments move up to different distances on the gel depending on their charge to mass ratio.
direct current 직류

The heavier fragments move a little, while the lighter DNA fragments move up to a larger distance. Following the migration of the molecules, the gel is treated with selective stains to show the location of separated molecules in the form of bands.
migration 이동
treat 다루다
selective stain 선택염색
Very large DNA molecules or chromosomes cannot be separated even by Agarose Gel electrophoresis. For separation of such very large DNA molecules (sometimes representing whole chromosomes), a new technique is used which is known as Pulse Field Gel Electrophoresis (PFGE).
separation 분리
Blotting Techniques:
Visualization of a specific DNA (or RNA or protein) fragment out of many molecules requires a technique called blot transfer. In this technique, the separated bands are transferred onto a nitrocellulose membrane from the gel.
Visualization 시각화
Mainly there are three types of blot transfer procedures:
Southern Blotting, Northern Blotting and Western blotting.
Southern blotting is named after the person who devised this technique, viz. E.M. Southern (1975). The other names began as laboratory jargon but they are now accepted terms.
procedure 절차
Technically, blotting may be defined as the transfer of macromolecules from the gel onto the surface of an immobilizing membrane like nitrocellulose membrane. It is to note here that during such transfer, the relative positions of bands (of macromolecules) are same on the membrane as they occurred on the gel.
macromolecule 거대분자
immobilize 고정시키다
The membranes which may.be used in blotting are nitrocellulose membrane, nylon membrane, carboxymethyl membrane, diazobenzyl-oxymethyl (DBM) membranes, etc.
Southern blotting is used for the transfer of DNA from gel onto the membrane while Northern and Western blotting are used for the transfer of RNA and protein bands respectively. One other blotting technique is south-western blotting which examines the protein-DNA interactions.
A schematic representation of southern blotting technique is given in the fig. 7. In this technique first of all, the sample DNA is digested with restriction enzymes to obtain fragments of different lengths. These differently sized DNA segments are then passed through Agarose Gel Electrophoresis for their separation based on their lengths.

The gel so obtained with different bands of DNA fragments is placed on top of buffer saturated filter papers which act as a filter paper wick. Above gel is put a nitrocellulose filter and over nitrocellulose filter are placed many dry filter paper sheets. With the movement of buffer towards the dry filter papers, the DNA bands are also moved upwards and hence they get bound to the nitrocellulose filter membrane.
Now, the nitrocellulose filter is removed and baked in vacuum. DNA fragments on the nitrocellulose filter are hybridized with single stranded radioactively labeled probes. Washing is done to remove unbound probes and finally the DNA bands with radioactivity are visualized by autoradiography.
In Northern Blotting, RNA molecules are blot transferred from the gel onto a chemically reactive paper. Western blotting is used for proteins and its working is based on the specificity of antibody-antigen reaction. In this technique the hybridization of bound proteins is done with radioactively labeled antibodies.
Dot Blot Hybridization:
The procedure of this technique is almost the same as blotting, but the only difference is that the DNA fragments are not separated by electrophoresis, instead they are directly applied as a dot on the nitrocellulose membrane.
Then radioactively labeled DNA probes having the complementary base sequences to the DNA of interest are applied on this membrane to allow its hybridization. The position of this hybridization is then detected by autoradiography method.
DNA Sequencing:
The segments of specific DNA molecules obtained by recombinant DNA technology can be analysed for determining their nucleotide sequence.
The methods commonly used for DNA sequencing are:
i. Enzymatic method or Sanger’s Dideoxy method.
ii. Chemical method or Maxam-Gilbert Method.
iii. Automated method.
(i) Enzymatic method of DNA sequencing is also called as Sanger-Coulson method of sequencing of DNA molecules. This method involves the use of single stranded DNA as a template for DNA synthesis.
The dideoxynucleotide triphosphates (ddNTPs like ddCTP, ddGTP, ddATP, ddTTP) are incorporated in the growing chain and they terminate the chain synthesis because they are unable to form a phosphodiester bond with next deoxy-nucleotide triphosphate.
For sequencing, the reaction mixture is taken in four separate test tubes. In each test tube is added one particular ddNTP. As a result, different sizes of newly synthesized DNA strands are obtained in each test tube which are terminated by a particular ddNTP. These segments are then separated by electrophoresis and then the DNA sequences are obtained by reading the bands on autoradiogram from bottom to the top of gel.
(ii) Chemical method of DNA sequencing involves the degradation of DNA by using chemicals, rather than synthesis of new DNA. In this type of sequencing, the DNA sample is labeled radioactively at 3′ ends and separated into single strands. Sample is then divided into four test tubes, each treated with a specific chemical reagent which degrades only at specific nucleotide base like G or C or ‘A and G’ or ‘C and T’.
As a result of this partial chemical cleavage, a number of differently sized fragments are obtained in each test tube. These fragments are separated by gel electrophoresis and then observed under autoradiography to interpret the nucleotide sequence of sample DNA. Chemical method is not used very Commonly because it is a slow and labour intensive process.
(iii) Automatic DNA sequencing methods have been developed by improvements in dideoxy-method. A number of automatic DNA sequencing machines have also been invented which are capable of sequencing thousands of nucleotides within few hours.
Such methods involve the tagging of fluorescent dyes to ddNTPs, slab gel sequencing systems, capillary gel sequencing systems and PCR-based DNA sequencing techniques. Such techniques are faster and more reliable.
Artificial Gene Synthesis:
This technique may also be called as oligonucleotide synthesis. It is one of those techniques which have been adopted for the synthesis of desired gene or DNA fragment. Gene synthesis is now a routine laboratory procedure to be utilized in the rec DNA technology.
synthesis 합성
First success in the approach of artificial gene synthesis was achieved by Dr. Har Govind Khorana and his co-workers in 1970 when they synthesized the artificial gene for a t-RNA in vitro which had potential for functioning within a living cell.
Major approaches available for the artificial synthesis of genes are:
Enzymatic synthesis of Gene:
artificial synthesis 인공합성
Enzymatic synthesis 효소적 합성
When details of base sequence of concerned gene are available, the polynucleotide of that same base sequence can be synthesized by enzymatic method. In this method the bacterial enzyme Polynucleotide phosphorylase is utilized. This method is easy to perform and does not require any template.
utilize 활용하다
Chemical synthesis of Gene:
Once the base sequence of a gene is deducted, this gene can be synthesized by a purely chemical method as used by Khorana and his co-workers for the synthesis of gene for yeast alanyl t-RNA. This method utilizes different chemical reagents for various steps of the process.
There are mainly three distinct methods, which are phosphodiester phosphotriester, and phosphite-triester methods. These methods differ in their strategies for protecting the hydroxyl group of the phosphate residues.
If the detailed sequence of the concerned gene is unknown then the artificial gene is synthesized in the form of cDNA i.e. complementary DNA from the mRNA of that gene. In this method, the enzyme employed is RNA directed DNA polymerase.
PCR (Polymerase Chain Reaction):
PCR is a technique for the amplification (or cloning) of a target sequence of DNA. It is sometimes also referred to as in vitro gene cloning (without expression of that gene). PCR is an important technique in molecular biology and it was discovered by Kary Mullis in 1985 (Fig. 8). It is carried out in vitro and by it, upto billion copies of the target DNA sequence can be obtained from a single copy within few hours only.
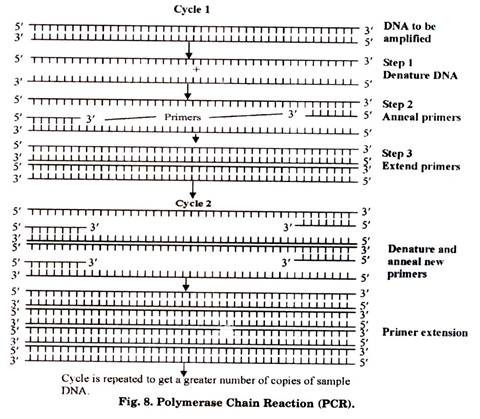
Outline of PCR:
PCR is a technique which results in selective amplification of a selected DNA molecule. One limitation of PCR is that the border region sequences of the DNA (to be amplified) must be known in order to select the appropriate primers which anneal (attach) at its 3′ ends. Primer annealing is important due to the fact that enzyme DNA polymerases require double stranded (ds) primer regions for initiating the DNA synthesis.
The whole reaction of PCR takes place in a tube called eppendorf tube. Scientists are using PCR in a number of disciplines due to the advantages like it is a quick, simple and extremely accurate technique. Major limitation of PCR is that due to its extreme sensitivity it may produce erroneous results caused by several inhibitors or contaminating DNA segments present in the sample DNA preparation.
Main Requirements of PCR:
a. Two nucleotide primers which are complementary to 3′ ends of target DNA strands
b. Target DNA sequence.
c. A heat stable DNA polymerase e.g. Taq polymerase.
d. Deoxy adenosine triphosphate (dATP)
e. Deoxy thymidine triphosphate (dTTP)
f. Deoxy cytidine triphosphate (dCTP)
g. Deoxy guanosine triphosphate (dGTP)
h. A thermal cycler in which PCR is carried out
Steps in PCR:
A generalized PCR-protocol involves following steps (however, the temperature-time profile may vary according to the requirements):
(a) Mix target DNA sequence, excess of primers, dATP, dTTP, dCTP, dGTP and Taq polymerase in the reaction mixture in eppendorf tube. Place this tube in thermal cycler.
(b) Reaction mixture is given high temperature of about 90-98°C for few seconds to denature the DNA. As a result the double stranded DNA becomes single stranded.
(c) Temperature is changed to about 55°C for 20 seconds so that primers are annealed at 3′ ends of DNA.
(d) Now the temperature is maintained at 72°C for 30 seconds which facilitates the functioning of Taq polymerase thus synthesizing the complementary strand of DNA.
(e) Hence, one cycle of PCR is completed here resulting in the formation of two ds DNA molecules from one ds DNA.
(f) Same cycle is repeated till the required number of DNA copies are obtained.
Main Types of PCR:
1. Inverse PCR
2. Anchored PCR
3. Asymmetric PCR
4. Overlap-Extension PCR
Uses of PCR:
1. For amplification of DNA
2. To detect mutations
3. To diagnose genetic disorders
4. To produce in vitro mutations
5. For preparing DNA for sequencing
6. To analyse genetic defects in single cells from human embryos.
7. To identify virus & bacteria in infectious diseases.
8. For characterization of genotypes.
Colony Hybridization Technique:
This technique is used in genetic engineering for the identification of transformed bacterial cells (i.e. cells which contain foreign DNA). After transformation of cells with a specific DNA, it is likely that only some of those cells may have foreign DNA. For further procedure, firstly it is important to screen such cells which are having foreign DNA.
This screening is done by using the technique of colony hybridization in case of bacterial cells (Fig. 9). A similar technique namely Plaque Hybridization is utilized for screening of transformed bacteriophages.
Basic principle of this technique lies in the in-situ hybridization of transformed bacterial cells with a radioactive probe sequence. Due to the specificity of probe, it enables rapid identification of one colony (through radioactivity) even amongst many thousands of colonies.
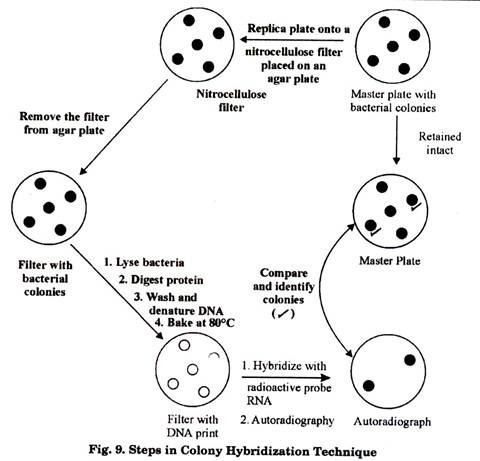
The transformed bacterial cells are first of all plated on a suitable agar plate which is termed as the master plate. Colonies are grown in the master plate. These colonies on the master plate are replica-plated onto a nitrocellulose or nylon membrane by placing it gently over the master plate. This replica-plate carrying the colonies is removed and treated with alkaline reagent to lyse the bacteria.
DNA of those bacterial cells is denatured. Proteins on the membrane are digested. Finally the membrane is washed to remove all other molecules, leaving behind only the denatured DNA bound to it, in the form of DNA print of the colonies.
This DNA print is then hybridized with a radioactively labeled RNA/DNA probe. Membrane is washed to remove any unbound probe and then autoradiography is done to detect radioactivity. The positions of the DNA prints showing up in autoradiograph are then compared with the master plate to identify the transformed colony.