TLR Signaling Pathways
Recognition of PAMPs by TLRs leads to transcriptional upregulation of distinct genes, depending on the TLRs and cell types involved (Figure 1). The difference in the signaling cascades activated by the individual TLRs can be partly explained by the TIR domain-containing adaptor molecules recruited to TLRs (Akira et al., 2006).
There are five TIR domain-containing adaptors including MyD88,TIR domain-containing adaptor inducing IFN-β (TRIF; also known as TICAM-1), TIRAP/Mal, TRIF-related adaptor molecule (TRAM), and Sterile-alpha and Armadillo motif-containing protein (SARM). TLR signaling is roughly divided into two distinct pathways depending on the usage of the distinct adaptor molecules, MyD88 and TRIF.
The MyD88-Dependent Signaling Pathway
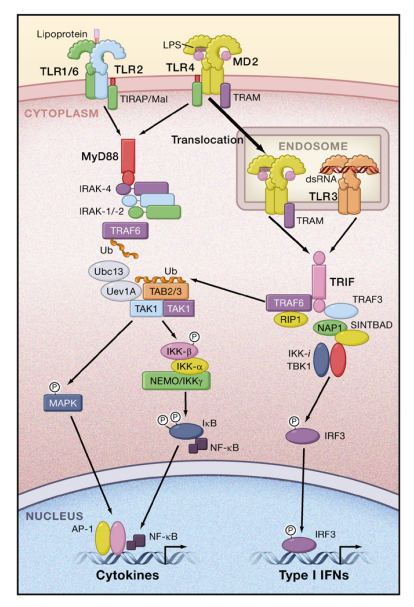
MyD88 is composed of a death domain (DD) in addition to a TIR domain. MyD88 is essential for the downstream signaling of various TLRs, with the exception of TLR3. Children with MyD88 deficiency suffer from recurrent pyogenic bacterial infections. TLR2 and TLR4 signaling requires TIRAP/Mal for bridging between TLR and MyD88.
MyD88 interacts with IL-1R-associated kinase (IRAK)-4, a serine/threonine kinase with an N-terminal death domain. IRAK-4 activates other IRAK family members, IRAK-1 and IRAK-2 (Kawagoe et al., 2008).
The IRAKs then dissociate from MyD88 and interact with TNFR-associated factor 6 (TRAF6), which acts as an E3 ubiquitin protein ligase. Together with an E2 ubiquitin-conjugating enzyme complex comprising Ubc13 and Uev1A, TRAF6 catalyzes the formation of a lysine 63 (K63)-linked polyubiquitin chain on TRAF6 itself as well as the generation of an unconjugated free polyubiquitin chain (Xia et al., 2009).
A complex of TGF-b-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TAB3 is activated by the unconjugated free K63 polyubiquitin chain and phosphorylates IkB kinase (IKK)-b and MAP kinase kinase 6. Subsequently, the IKK complex, composed of IKK-a, IKK-b, and NF-kB essential modulator (NEMO), phosphorylates IkBa, an NF-kB inhibitory protein.
Phosphorylated IkB undergoes degradation by the ubiquitin-proteasome system, thereby freeing NF-kB to translocate into the nucleus and activate expression of proinflammatory cytokine genes. Activation of the MAP kinase cascade is responsible for the formation of another transcription factor complex, AP-1, that targets cytokine genes.
TLR7 and TLR9 signaling induces the production of type I IFNs in addition to other NF-kB-dependent cytokines in a MyD88- dependent manner. In pDCs, MyD88 forms a complex with IRAK-1, TRAF6, TRAF3, IKK-a, and IRF7, and phosphorylated IRF7 translocates to the nucleus to activate the expression of genes encoding type I IFNs (Figure 2). In cDCs, IRF1, but not IRF7, is activated downstream of TLR7 and TLR9, resulting in the activation of IFN-b gene expression (Negishi et al., 2006; Schmitz et al., 2007).
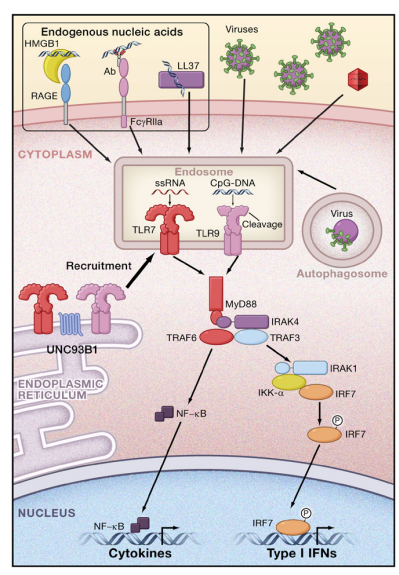
The TRIF-Dependent Signaling Pathway
In response to stimulation with dsRNA, TLR3 recruits another adaptor protein, TRIF. TLR4 triggers both MyD88-dependent and TRIF-dependent signaling. TLR4, but not TLR3, requires another adaptor, TRAM, for activating TRIF. A splice variant of TRAM called the TRAM adaptor with GOLD domain (TAG) acts as the negative regulator of TRIF-dependent signaling (Palsson-McDermott et al., 2009).
TRIF associates with TRAF3 and TRAF6 through TRAF-binding motifs present in its N-terminal portion. TRIF also contains a C-terminal receptor-interacting protein (RIP) homotypic interaction motif (RHIM) and interacts with RIP1 and RIP3 via this motif. In humans, SARM functions as an inhibitor of TRIF-dependent signaling (Carty et al., 2006).
The TNFR-associated death domain protein (TRADD), an essential adaptor for TNFR signaling, is involved in the TRIF-dependent signaling pathway (Ermolaeva et al., 2008; Pobezinskaya et al., 2008). TRADD forms a complex with FAS-associated death domain-containing protein (FADD) and RIP1, and TRADD mediates ubiquitination of RIP1, an event required for NF-kB activation. FADD activates caspase-8 or caspase-10 in response to poly I:C, and the cleaved form of caspases activates NF-kB (Takahashi et al., 2006). TRAF3 is important for activating two IKK-related kinases, TANK-binding kinase 1 (TBK1) and IKK-i (also known as IKK-3) (Hacker et al., 2006; Oganesyan et al., 2006).
TRAF3 undergoes K63-linked auto-ubiquitination in response to TLR3 and acts as an E3 ubiquitin ligase. TRAF3 activation is negatively regulated by a deubiquitination enzyme DUBA (Kayagaki et al., 2007), and MyD88-dependent signaling triggers K48-linked ubiquitination of TRAF3. Proteasome-mediated degradation of TRAF3 is important for the activation of MAP kinases and the production of proinflammatory cytokines (Tseng et al., 2009). A recent study identified an E2 ubiquitin ligase, Ubc5, as a molecule required for IRF3 activation by catalyzing K63-type polyubiquitin chain formation (Zeng et al., 2009). TBK1 and IKK-i phosphorylate IRF3 and IRF7; IRF3 and IRF7 dimers translocate to the nucleus, resulting in induction of type I IFNs and expression of IFN-inducible genes. IKK-i also phosphorylates STAT1 to facilitate the induction of a set of IFN-inducible genes including Adar1, Ifit3, and Irf7 (Tenoever et al., 2007).
The activation of TBK1 and IKK-i is modulated by various proteins. TBK1 and IKK-i interact with TRAF family memberassociated NF-kB activator (TANK) (also known as I-TRAF), NAK-associated protein 1 (NAP1), and the TBK1 adaptor (SINTBAD), which is similar to NAP1 (Guo and Cheng, 2007; Ryzhakov and Randow, 2007; Sasai et al., 2006). These molecules contain a TBK1-binding motif and show similarities in their coiled-coil domains. However, TANK/ cells do not show impaired type I IFN production in response to dsRNA stimulation (Kawagoe et al., 2009). Although knockdown of either NAP1 or SINTBAD impairs TRIF signaling, the relationship between these molecules in TRIF signaling is not yet fully understood.
RLRs and Virus Recognition
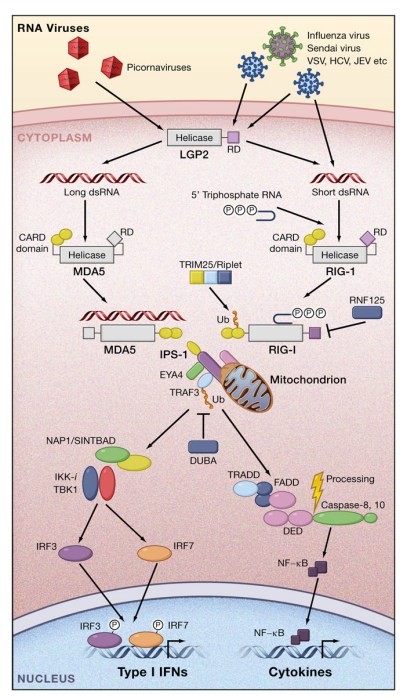
Figure 3. Recognition of RNA Viruses by RLRs RIG-I and MDA5 recognize different RNA viruses by detecting short dsRNAs with 50 triphosphate ends and long dsRNAs, respectively. LGP2 functions as a positive regulator in RIG-I-mediated and MDA5-mediated virus recognition. Activation of RIG-I is positively and negatively regulated by ubiquitin ligases TRIM25 and RNF125, respectively. RIG-I and MDA5 interact with IPS-1 through homophilic interactions between CARD domains. IPS-1 then activates signaling cascades leading to the expression of type I IFN genes via EYA4, TRAF3, NAP1/SINTBAD, TBK1/IKK-i, and IRF3/7. RLR signaling mediates polyubiquitination of TRAF3, which is removed by a deubiquitinase, DUBA. Simultaneously, IPS-1 signaling induces nuclear translocation of NF-kB via TRADD and FADD and caspase-8/-10. A cleaved fragment of caspase-8/-10 is responsible for the activation of NF-kB.
The RIG-I-like receptor (RLR) family is composed of RIG-I, melanoma differentiation-associated gene 5 (MDA5), and LGP2 (Takeuchi and Akira, 2009; Yoneyama and Fujita, 2008). RLRs are composed of two N-terminal caspase recruitment domains (CARDs), a central DEAD box helicase/ATPase domain, and a C-terminal regulatory domain. They are localized in the cytoplasm and recognize the genomic RNA of dsRNA viruses and dsRNA generated as the replication intermediate of ssRNA viruses.
The expression of RLRs is greatly enhanced in response to type I IFN stimulation or virus infection. Mouse fibroblasts and cDCs lacking RIG-I are defective in the production of type I IFNs and inflammatory cytokines in response to various RNA viruses (Kato et al., 2006). These include the Paramyxoviridae such as Newcastle disease virus (NDV) and Sendai virus (SeV), vesicular stomatitis virus (VSV), influenza virus, and Japanese encephalitis virus (JEV). In contrast, cells lacking MDA5 respond normally to these viruses. Meanwhile, the IFN responses to several Picornaviridae, including encephalomyocarditis virus (EMCV), Mengo virus, and Theiler’s virus, are abrogated in MDA5/ cells, but not in cells lacking RIG-I. In addition to JEV, another flavivirus, hepatitis C virus (HCV), is also recognized by RIG-I, whereas both RIG-I and MDA5 redundantly recognize Dengue virus and West Nile virus (Loo et al., 2008; Sumpter et al., 2005). A double-stranded segmented RNA virus, reovirus, induces IFN production mainly through MDA5. However, the absence of both RIG-I and MDA5 completely abrogates this IFN production, suggesting that both RIG-I and MDA5 are involved in the recognition of reovirus (Kato et al., 2008; Loo et al., 2008). Mouse embryonic fibroblasts (MEFs) derived from RIG-I/MDA5/ mice failed to produce type I IFNs to any of the RNA viruses tested, indicating that RIG-I and MDA5 are essential and sufficient for evoking type I IFN production in response to RNA viruses. RIG-I recognizes relatively short dsRNA (up to 1 kb), and the presence of a 50 triphosphate end greatly enhances its type I IFN-inducing activity (Figure 3). It has been postulated that 50 triphosphate ssRNA synthesized by in vitro transcription is a RIG-I ligand (Hornung et al., 2006; Pichlmair et al., 2006). However, recent studies have shown that T7 polymerase commonly produces extended byproducts generating dsRNA (Schlee et al., 2009; Schmidt et al., 2009). Chemically synthesized 50 triphosphate ssRNA failed to stimulate RIG-I, indicating that RNA needs to be double-stranded for activation of RIG-I. Regarding a minimum length, the 19-mer or 21-mer dsRNA with a 50 triphosphate end is able to potently induce type I IFNs. In contrast, a 50 triphosphate end is not always necessary, as chemically synthesized dsRNAs with a 50 monophosphate end or those without a 50 phosphate can potently activate RIG-I (Takahasi et al., 2008; Kato et al., 2008). The amount of IFNs produced by these RNAs is low compared to dsRNA with a 50 triphosphate end. HCV genomic RNA has been screened for RNA sequences that activate RIG-I. Poly(U)- or poly(A)-rich sequences from the HCV RNA 30 untranslated region (UTR) are responsible for RIG-I-mediated IFN production, based on RNAs generated by in vitro transcription using T7 polymerase (Saito et al., 2008). However, in vitro-transcribed RNAs with low U/A content also potently activate RIG-I. Therefore, further studies are required to clarify whether particular RNA sequences are important for RIG-I-mediated recognition. VSV produces dsRNA in infected cells (Kato et al., 2008). Disruption of dsRNA among RNAs from VSV-infected cells reduces the IFN-b-inducing activity, suggesting that the presence of dsRNA in VSV-infected cells is important for recognition by RIG-I. Interestingly, the dsRNA fragments produced by VSV infection are about 2.0–2.5 kb, and much shorter than the size of the VSV genomic RNA. It has been reported that defective interfering (DI) particles are generated in VSV-infected cells, and that the sizes of DI particle dsRNAs are about 2.2 kb (Pattnaik et al., 1995). Thus, the dsRNA generated during the course of VSV replication may be derived from DI particles, although further studies are needed to clarify the source of this dsRNA. As DI particles are known to strongly induce type I IFNs, RIG-I may play a role in detecting the presence of dsRNA in DI particles. In contrast, it is difficult to detect dsRNA in cells infected with influenza virus. Genomic RNA from influenza virus lost its IFN-inducing activity after a phosphatase treatment that removed the 50 triphosphate end. The sequences of the 50 and 30 ends of the viral RNA are partially complementary to each other, and it has been suggested that a panhandle structure can form (Hsu et al., 1987). Thus, it is tempting to speculate that short panhandle dsRNA with a 50 triphosphate is responsible for recognition by RIG-I. In contrast to RIG-I, MDA5 detects long dsRNA (more than 2 kb) such as poly I:C. MDA5/ mice show severely reduced production of type I IFNs in response to poly I:C inoculation in vivo, whereas their production of IL-12p40 is not impaired (Kato et al., 2006). Shortening the length of the poly I:C by treatment with a dsRNA-specific nuclease converts poly I:C from an MDA5 ligand to a RIG-I ligand, indicating that long, but not short, dsRNA is recognized by MDA5. EMCV produces high levels of dsRNA in infected cells, and the 50 end of its genomic RNA is covalently linked to a small protein, VPg. Investigation of RNA from EMCV-stimulated cells has revealed that higher-order structure RNA containing both dsRNA and ssRNA, but not simple dsRNA as the replication intermediate, has MDA5-stimulating activity (Pichlmair et al., 2009). LGP2, the third member of the RLR family, lacks a CARD, and in vitro studies have suggested that LGP2 functions as a negative regulator of RIG-I and MDA5 responses by sequestering dsRNA or inhibiting RIG-I conformational changes (Rothenfusser et al., 2005; Saito et al., 2007; Yoneyama et al., 2005). However, the generation of LGP2/ mice and mice with a point mutation, D30A, that disrupts the ATPase activity of LGP2 revealed that LGP2 positively regulates production of type I IFNs in response to RNA viruses recognized by both RIG-I and MDA5 (Satoh et al., 2010). Nevertheless, LGP2 is dispensable for type I IFN production following transfection by synthetic RNAs. These results suggest that LPS2 may modify viral RNA by removing proteins from viral ribonucleoprotein (RNP) complexes or unwinding complex RNA structures to facilitate MDA5-mediated and RIG-I-mediated recognition of dsRNA. RLRs contain a C-terminal regulatory domain, which is responsible for the binding to dsRNAs. The recent solution of RLR C-terminal regulatory domain structures revealed that the C-terminal domains of LGP2 and RIG-I have a large basic surface forming an RNA-binding loop (Cui et al., 2008; Takahasi et al., 2008, 2009). The RIG-I and LGP2 C-terminal domains bind to the termini of dsRNA. Although the MDA5 C-terminal domain also has a large basic surface, it is extensively flat because of the open conformation of the RNA-binding loop. Therefore, the RNA-binding activity of MDA5 is much weaker than that of RIG-I and LGP2. RLRs catalyze ATP via the DExD/H helicase domain, and the ATPase activity is essential for RLRs to induce type I IFN production. Although RIG-I has helicase activity, it is not clear if the unwinding of dsRNA by RIG-I is required for triggering the signaling pathway. RIG-I might change its conformation to expose the CARDs and dimerize by catalyzing ATP, or alternatively, RIG-I may act as a translocase for dsRNA.
The RLR Signaling Pathways
The RIG-I conformation is known to be modulated by ubiquitination. TRIM25 and Riplet (also known as RNF135) act as E3 ubiquitin ligases that mediate the K63-linked polyubiquitination of RIG-I (Gack et al., 2007; Pichlmair et al., 2009). This modification is required for the activation of RLR signaling. On the other hand, K48-type polyubiquitination of RIG-I by RNF125 leads to RIG-I degradation by the proteasome and inhibition of RIG-I signaling (Arimoto et al., 2007). The CARDs of RLRs are responsible for triggering signaling cascades by interacting with the N-terminal CARD-containing adaptor IFN-b-promoter stimulator 1 (IPS-1) (also known as MAVS, CARDIF, or VISA) (Kawai and Akira, 2006) (Figure 3). IPS-1 is localized on the mitochondrial membrane, and cleavage of IPS-1 by an HCV NS3/4A protease, which dislodges it from the mitochondrial membrane, results in abrogation of RLR signaling. NLRX1 (also known as NOD9), an NLR family member, is localized on the mitochondrial membrane and acts as an inhibitor of IPS-1 signaling (Moore et al., 2008). However, another report showed that NLRX1 is a mitochondrial matrix protein responsible for the generation of reactive oxygen species. Therefore, further studies are required to clarify the role of NLRX1 in RLR signaling (Arnoult et al., 2009). IPS-1 activates TRAF3 and TRADD (Michallet et al., 2008). The downstream signaling molecules for the expression of IFN-inducible genes are shared between the IPS-1 and TRIF signaling pathways. The recently identified protein Eyes absent 4 (EYA4) associates with IPS-1 and NLRX1 and stimulates the expression of IFN-inducible genes in response to DNA stimulation (Okabe et al., 2009). EYA4 is a phosphatase for both phosphotyrosine and phosphothreonine, and the threonine phosphatase activity is essential for enhancing IFN-b gene activation. Identification of molecules that are dephosphorylated by EYA4 will be important for understanding the mechanism for how EYA4 regulates IFN responses. In contrast to pDCs, autophagy negatively regulates IFN responses in fibroblasts and cDCs by suppressing RLR signaling (Jounai et al., 2007; Tal et al., 2009). In the absence of autophagy-related proteins, such as ATG5 or ATG16L1, damaged mitochondria accumulate together with IPS-1, and reactive oxygen species associated with dysfunctional mitochondria are responsible for the overproduction of type I IFNs.
Recognition of Cytoplasmic DNA
Although DNA with a CpG motif is sensed by TLR9, introduction of dsDNA into cells evokes type I IFN responses in a TLR9- independent manner. Right-handed spiral B-DNA, but not lefthanded Z-DNA, activates these responses (Ishii et al., 2006). Although the sequence specificity has not been clearly observed, synthetic poly (dA:dT) is more potent for induction of IFN responses than poly (dI:dC) or poly (dG:dC). Transfection of dsDNA leads to the activation of IRF3 via TBK1 and IKK-i, but activation of NF-kB is barely detected in response to DNA. It is believed that cytoplasmic DNA recognition is important for the production of type I IFNs to infection with DNA viruses. Infection with intracellular bacteria such as Listeria monocytogenes and Legionella pneumophila induces type I IFN production in response to bacterial DNA in the cytoplasm (Stetson and Medzhitov, 2006). In addition, the adjuvant effect of DNA vaccines can be explained by TLR9-independent cytoplasmic recognition of DNAs that activates the TBK1-IRF3-dependent pathway (Ishii et al., 2008). Two studies have shown that poly (dA:dT) can be transcribed into dsRNA by polymerase III and that dsRNA is recognized in a RIG-I-IPS-1-dependent manner in human cells or transformed mouse MEFs (Figure 4) (Ablasser et al., 2009; Chiu et al., 2009; Choi et al., 2009). However, primary DCs prepared from RIG-Ideficient mice do not show a defect in IFN production, suggesting that cytoplasmic DNAs are sensed in both a polymerase III-RIG-I-dependent and -independent manner depending on the species or cell types involved. A cytoplasmic DNA-binding protein, DNA-dependent activator of IRF (DAI, also known as Zbp1 or DLM1), interacts with TBK1 and activates type I IFN responses (Takaoka et al., 2007). Nevertheless, Zbp1/ MEFs are still capable of producing type I IFNs in response to cytoplasmic DNA, suggesting that cytoplasmic DNA is redundantly recognized by as-yet unidentified receptors (Ishii et al., 2008). Expression cloning of a gene inducing IFN-b promoter activation identified STING (also known as MITA, ERIS, or TMEM173), a transmembrane protein expressed in the ER (Ishikawa and Barber, 2008; Sun et al., 2009; Zhong et al., 2008). Cells and mice lacking STING show impaired IFN production in response to both RNA and DNA stimulation. STING associates with a member of the translocon-associated protein (TRAP) complex that is required for protein translocation across the ER in resting cells. In response to transfection of dsDNA, STING translocates from the ER to the Golgi apparatus and subsequently to cytoplasmic punctate structures where it colocalizes with TBK1 (Ishikawa et al., 2009). The exocyst complex is involved in secretory vesicle sorting and is especially required for post-Golgi transport to the plasma membrane. Sec5, a component of the exocyst complex, colocalizes with STING. Activation of the RalB GTPase promotes the assembly of TBK1 and Sec5, leading to phosphorylation of Sec5 by TBK1. STING/ mice are highly susceptible to HSV-1 or VSV infection, indicating a role for STING in host defense against virus infection. Interestingly, autophagy-related gene 9a (ATG9a) colocalizes with STING (Saitoh et al., 2009). Loss of ATG9a, but not another autophagy-related gene (ATG7), greatly enhances the assembly of STING with TBK1 in response to dsDNA, followed by increased production of type I IFNs. Notably, the introduction of dsRNA did not induce the translocation of STING; it will be interesting to explore how STING regulates the IFN response to dsRNA. It has been shown that TBK1 activation by RalB is involved in cell transformation and tumor progression (Chien et al., 2006). Furthermore, a recent study revealed that TBK1 is required for KRAS-driven cancer formation, probably by activating the NF-kB-induced antiapoptotic gene BCL-xL (Barbie et al., 2009). Thus, TBK1 is a pleiotropic kinase involved in both innate immunity and tumorigenesis.
High-mobility group box (HMGB) proteins were originally identified as nuclear proteins with DNA-binding capacity. HMGB1 is also known to be secreted in response to cell damage and evokes inflammatory responses. Recently, HMGB proteins present in the cytoplasm were found to act as the initial sensors for cytoplasmic nucleic acids that lead to the activation of downstream receptors such as RLRs, TLRs, and unknown DNA receptors (Yanai et al., 2009). Although stimulation with TLR ligands alone does not lead to activation of the inflammasome, the introduction of dsDNA induces not only the production of pro-IL-b but also its cleavage via caspase-1. Sensing of cytoplasmic DNA by absent-in-melanoma 2 (AIM2) activates the inflammasome via ASC and caspase-1, leading to the production of IL-1b (see Review by K. Schroder and J. Tschopp on page 821).
NLR- and CLR-Mediated Pathogen Recognition
The NLR family consists of cytoplasmic pathogen sensors that are composed of a central nucleotide-binding domain and C-terminal leucine-rich repeats (Inohara et al., 2005). The N-terminal portions of most NLRs harbor protein-binding motifs, such as CARDs, a pyrin domain, and a baculovirus inhibitor of apoptosis protein repeat (BIR) domain. NLRs harboring a pyrin domain or a BIR domain in their N terminus are not involved in the transcriptional activation of inflammatory mediators and are components of the inflammasome that regulates caspase1 activation. NOD1 and NOD2, which harbor CARDs in addition to NOD and LRR domains, activate NF-kB via an adaptor, RIP2/ RICK. NOD1 and NOD2 induce transcriptional upregulation of proinflammatory cytokine genes. NOD1 and NOD2 recognize the structures of bacterial peptidoglycans, g-D-glutamyl-mesodiaminopimelic acid (iE-DAP) and muramyl dipeptide (MDP), respectively. As TLRs also recognize bacterial peptidoglycan components, TLRs and NODs synergistically activate proinflammatory cytokine production. In addition to bacteria, a recent study found that the expression of NOD2 is involved in 50 -triphosphate RNA-induced type I IFN production and host defense against respiratory syncytial virus infection (Sabbah et al., 2009). CLRs comprise a transmembrane receptor family characterized by the presence of a carbohydrate-binding domain. CLRs recognize carbohydrates on microorganisms such as viruses, bacteria, and fungi. CLRs either stimulate the production of proinflammatory cytokines or inhibit TLR-mediated immune complexes. The functions of CLRs are described in detail elsewhere (Geijtenbeek and Gringhuis, 2009), and here we give just a few examples of CLR-mediated microbial recognition. Dectin-1 and dectin-2 are immunoreceptor tyrosine-based activation motif (ITAM)-coupled CLRs responsible for sensing b-glucans from fungi. DCs activated by dectin-1 or dectin-2 are able to instruct T cells to confer protective immunity against Candida albicans (Robinson et al., 2009). The macrophage C-type lectin MINCLE (also known as Clec4e and Clecsf9) senses infection by fungi such as Malassezia and Candida (Yamasaki et al., 2009). In addition, MINCLE is responsible for the detection of an endogenous protein, spliceosome-associated protein 130 (SAP130), which is a component of U2 snRNP from necrotic host cells (Yamasaki et al., 2008). CLEC9A is expressed on CD8+ DCs in the spleen and also recognizes necrotic cells for cross-priming. CLRs activate intracellular signaling either via the ITAM domain of CLRs or via adaptors harboring an ITAM domain, such as FcRg, DAP10, or DAP12. The Syk tyrosine kinase is activated by ITAM-containing proteins either directly or indirectly via ITAM-containing adaptors. Engagement of Syk with CLRs activates MAP kinases, the transcription factor NF-AT, and NF-kB through CARD9. As a result, proinflammatory cytokines are produced. However, the signaling mechanism for the production of proinflammatory cytokines is not well understood.
Transcriptional Regulation of Inflammatory Mediators
Activation of PRR signaling pathways leads to the nuclear translocation of a set of transcription factors, including NF-kB, AP-1, IRFs, and C/EBPb. These factors cooperatively regulate the transcription of their target genes. Furthermore, remodeling of chromatin is important for controlling the transcriptional regulation of a set of TLR-inducible genes (Ramirez-Carrozzi et al., 2006). In addition, epigenetic controls of a set of TLR-inducible genes are critical for induction of tolerance to LPS, a well-known phenomenon rendering cells refractory to a second exposure to LPS (Foster et al., 2007) (see Review by S.T. Smale on page 833 of this issue). Here, we focus on the TLR-inducible proteins that modulate inflammatory responses. Besides cytokines, chemokines, and IFNs, TLR stimulation upregulates the expression of hundreds of genes in macrophages. These genes are potentially involved in antimicrobial defense (e.g., defensins, lipocaline), metabolic changes, and tissue repair (e.g., secretory leukocyte peptidase inhibitor). Nevertheless, the functions of various TLR-inducible genes remain largely unexplored. Some gene products are associated with the positive and negative regulation of inflammatory responses by controlling TLR-signaling pathways. The genes immediately induced by TLR ligands potentially regulate TLR signaling or responses after their expression. It is well known that the components of NF-kB are TLRinduced genes that undergo positive feedback regulation by NF-kB (Figure 5). In addition, IkBz and activating transcription factor 3 (ATF3) are nuclear factors that are rapidly induced by the TLRs (Gilchrist et al., 2006; Yamamoto et al., 2004). IkBz harbors C-terminal ankyrin repeat motifs that bind to the NF-kBp50 subunit, and expressed IkBz positively regulates the induction of a set of genes including IL-6 and IL-12p40, which are transcribed later. In contrast, ATF3 negatively regulates the transcription of genes encoding IL-6 and IL-12p40 by increasing histone deacetylase activity. Epigenetic changes can be regulated by TLR-inducible proteins. Trimethyl-histone 3 lysine 27 demethylase, Jmjd3, is expressed in response to TLR stimulation via NF-kB and is recruited to the transcription start sites of LPS-inducible genes (De Santa et al., 2007). Given that Jmjd3 is recruited to the transcription start site of various LPS-inducible genes, it may be responsible for the fine-tuning of LPS-induced gene expression in macrophages (De Santa et al., 2009). In response to TLR ligand stimulation, genes encoding RNA-binding proteins with a CCCH-type zinc finger motif (Zf) are rapidly induced. These proteins include Zc3h12a, Zc3h12c, Zc3hav1, and tristetraprolin (TTP; also known as Zfp36) (Matsushita et al., 2009). TTP harbors two CCCH-type Zf domains and associates with mRNA via AU-rich elements present in the 30 UTR, leading to removal of the poly(A) tail by recruitment of a deadenylase (Carrick et al., 2004). The deadenylated mRNAs traffic to exosomes where the RNA is degraded. TTP is required to prevent the generation of autoimmune arthritis in mice through controlling mRNAs encoding TNF. Zc3h12a and Zc3h12c are composed of a CCCH-type Zf domain and an RNase domain (Matsushita et al., 2009). Zc3h12a is responsible for degrading mRNAs for TLR-inducible proteins such as IL-6 and IL-12p40 in macrophages. Zc3h12a controls its target mRNAs via the 30 UTR independently of AU-rich elements; the RNase activity is essential for mRNA degradation. Zc3h12a/ mice spontaneously develop a fatal inflammatory disease characterized by highly elevated immunoglobulin levels and autoantibody production. RLR signaling induces another gene encoding a CCCH-type Zf protein, ZAP (also known as Zc3hav1). ZAP contains four clusters of CCCH-type Zf domains and directly binds to viral RNAs as well as exosomes where viral RNA is degraded (Gao et al., 2002). ZAP overexpression is reported to confer resistance against various viruses such as retroviruses and alphaviruses. Zcchc11 is a CCHC-type Zf domaincontaining RNA-binding protein with a nucleotidyltransferase domain. A recent study found that Zcchc11 adds terminal uridines to miR-26 family microRNAs, which target IL-6 mRNA (Jones et al., 2009). Uridylated miR-26 fails to repress IL-6 mRNA, and Zcchc11 thereby potentiates IL-6 production in response to TNF stimulation. It was reported that a set of mRNAs rapidly expressed in response to TNF stimulation are critically controlled through mRNA stability. These mRNAs tend to have abundant AU-rich elements in their 30 UTRs compared with mRNAs expressed at later time points (Hao and Baltimore, 2009). Therefore, control of mRNA decay may be as important as control of transcription in terms of the regulation of innate immune responses. TLR signaling induces the expression not only of proteincoding mRNAs but also of noncoding RNAs (Guttman et al., 2009). Some of the noncoding RNAs produce microRNAs including miR-146a/b, miR-147, and miR-155 (Taganov et al., 2006). These microRNAs interact with their target mRNAs and fine-tune their expression to modulate the inflammatory response. One of the best characterized microRNAs is miR-155, which is rapidly induced in response to TLR ligands and modulates innate and adaptive immune responses by modulating the expression of multiple target mRNAs encoding PU.1, IKK-i, SHIP1, and so on (Faraoni et al., 2009). A recent report shows that miR-21 negatively regulates TLR4 signaling by the tumor suppressor PDCD4, a protein required for NF-kB activation (Sheedy et al., 2009).
PRR-Dependent Recognition of Self-Nucleotides in Autoimmunity
Under physiological conditions, PRRs strictly discriminate self and microbial components to prevent the development of autoimmune disease. A hallmark of autoimmune disease is the production of antibodies recognizing self-antigens. In human autoimmune diseases such as systemic lupus erythematosus (SLE), immune complexes are formed by autoantibodies and deposited in the kidneys, causing glomerular nephritis. It has been shown that serum type I IFN levels in SLE patients positively correlate with disease severity. TLRs, especially TLR7 and TLR9, have been implicated in the production of autoantibodies in various mouse models. Selfnucleic acids released from damaged cells are rapidly degraded by serum nucleases and do not encounter TLR7 or TLR9. However, when self-nucleic acids interact with cellular proteins such as HMGB1, RNPs, antimicrobial peptides, and autoantibodies, endocytosis of the resulting nucleic acid-protein complexes is facilitated and induces TLR7- and TLR9-mediated type I IFN production (Figure 2). For instance, HMGB1 interacts with the receptor for advanced glycation end-products (RAGE) on the cell surface, and HMGB1-DNA-containing immune complexes released from damaged cells stimulate TLR9 and RAGE, which cooperate to stimulate pDCs and B cells (Tian et al., 2007). Endogenous RNA-protein complexes, such as U1 snRNP, can activate autoreactive B cells and DCs via TLR7 (Vollmer et al., 2005). Furthermore, nucleic acids bound to autoantibodies are endocytosed through B cell receptors and FcgRIIA in B cells and DCs (Means et al., 2005; Viglianti et al., 2003). Recognition of immune complexes by these receptors leads to the activation of autoreactive B cells as well as the production of type I IFNs, thus exacerbating the causes of autoimmune diseases. Similarly, the cationic antimicrobial peptide LL37 forms a complex with self-DNA or self-RNA, thereby facilitating endocytosis by pDCs and stimulating TLR-mediated IFN responses (Ganguly et al., 2009; Lande et al., 2007). LL37 is highly expressed in psoriasis skin lesions, and the production of IFNs contributes to the pathogenesis of this disease. Taken together these data show that the responses of nucleic acidsensing TLRs are positively regulated by endogenous proteins that facilitate endocytosis of nucleic acids. Type I IFNs and cytokines produced by the TLRs cause inflammation, and this positive feedback mechanism is involved in the exacerbation of autoimmune diseases. The contributions of nucleic acid-sensing TLRs to autoimmunity have been examined using mouse models that develop autoimmune disease spontaneously. Duplication of the Tlr7 gene accounts for the autoimmune phenotypes associated with Y chromosome-linked autoimmune accelerator (Yaa) mice (Pisitkun et al., 2006; Subramanian et al., 2006). Reciprocally, TLR7 deficiency in lupus-prone MRL lpr/lpr mice results in reduced levels of autoantibodies recognizing RNA-containing antigens and abrogation of disease (Christensen et al., 2006). But the relationship between TLR9 and autoimmune disease mouse models is complicated. Although CpG-DNA has been used as an adjuvant to generate organ-specific autoimmune disease in mouse models, the lack of TLR9 exacerbated the severity of disease in the MRL lpr/lpr murine lupus model. Although the TLR7 and TLR9 signaling pathways are indistinguishable, there is a clear difference in the roles of TLR7 and TLR9 in the establishment of systemic autoimmune disease. A mutation of amino acid D34 in UNC93B1 that changes its ability to bind to TLR7 and TLR9 has been identified (Fukui et al., 2009). The mutant UNC93B1 increases TLR7 responses but causes hyporesponsiveness to TLR9. Given that TLR7 is important for the generation of autoimmune disease models in mice, it is possible that UNC93B1 suppresses RNA sensing by skewing the response toward DNA. Appropriate clearance of nucleic acids is essential for preventing autoimmune disease. DNase II is located in the lysosomes of macrophages and is required for the degradation of DNA from apoptotic cells. In the absence of DNase II, undigested DNA accumulates in macrophages, and the resulting production of IFN-b and TNF causes embryonic lethality (Kawane et al., 2003). The type I IFNs are produced independently of TLR9, suggesting that recognition of DNA by intracellular receptors is responsible for the embryonic lethality of DNase II-deficient mice (Okabe et al., 2005). Similarly, deficiency in DNase I, a major DNase present in serum, results in the generation of SLE-like autoimmune disease in mice; a mutation in DNase I has been identified in human SLE patients (Lee-Kirsch et al., 2007). Aicardi-Goutie`res syndrome (AGS) is a human disorder characterized by fatal encephalopathy owing to overproduction of IFN-a. Recent studies have revealed that AGS is caused by a mutation in 30 repair exonuclease 1 (Trex1) (Crow et al., 2006). It has been shown that DNA derived from endogenous retroelements accumulates in Trex1/ cells and stimulates the induction of IFN-inducible genes (Stetson et al., 2008). In addition, mutations in components of RNase H2 are also involved in AGS (Rice et al., 2009). Nucleic acid accumulation because of a deficiency in these nucleases probably leads to recognition of the nucleic acids by cytoplasmic PRRs resulting in the production of type I IFNs. Interestingly, mutations in MDA5 that disrupt its signaling ability correlate with resistance to type I diabetes, although the mechanism for the contribution of MDA5 to this disease remains unclear (Nejentsev et al., 2009). Future studies will clarify how cytoplasmic PRRs contribute to the generation of autoimmune disease.
PRR Activation in Acute and Chronic Inflammation
PRRs recognizing non-nucleic acid ligands are also involved in various inflammatory and autoimmune diseases. It is well known that activation of TLR signaling by bacterial components leads to acute inflammation, culminating in septic shock in some cases. TLR ligands also are commonly used as adjuvants to generate organ-specific autoimmune disease models in mice for arthritis or encephalitis, for example. A mutation in NOD2 that generates a truncated form of the protein is frequently found in patients with Crohn’s disease, an inflammatory bowel disorder (Cho, 2008). This disease may arise through impairment of proper responses to intestinal bacteria and aberrant bacterial growth causing increased inflammatory responses in the intestine. Mutations in the autophagy-related gene ATG16L1 are also implicated in susceptibility to Crohn’s disease. In the absence of ATG16L1 in mice, overactivation of the inflammasome occurs, and increased production of IL-1b and IL-18 contributes to inflammation in the intestine (Saitoh et al., 2008). Furthermore, ATG16L1 is critical for the normal function of intestinal Paneth cells (Cadwell et al., 2008). Lack of negative regulators for TLRs can cause autoimmune disease based on various mouse models. Loss of the protein tyrosine phosphatase SHP1 in mice induced by N-ethyl-N-nitrosourea (ENU) mutagenesis leads to the generation of inflammatory lesions together with overactivation of macrophages in response to TLR stimulation (Croker et al., 2008). The inflammation is suppressed by loss of MyD88, suggesting that microbeinduced hyperactivation of TLR signaling is responsible for the generation of this inflammatory disease. A20 is a protein with dual enzyme domains, comprising an E3 ubiquitin ligase and a deubiquitinase that removes K63-linked polyubiquitin chains. A20 negatively regulates NF-kB activation downstream of TLR2 and NOD1/NOD2 (Boone et al., 2004; Hitotsumatsu et al., 2008). In the absence of A20, mice develop multiorgan inflammatory disorders that lead to premature death. Another example of a TLR negative regulator is TANK. Generation of mice lacking TANK revealed that TANK is critical for controlling TLR signaling in macrophages as well as antigen receptor signaling in B cells, by inhibiting the activation of TRAF6 (Kawagoe et al., 2009). TANK/ mice spontaneously develop autoimmune glomerular nephritis depending on the presence of MyD88 or IL-6. These results suggest that aberrant activation of innate immune cells and B cells is responsible for the generation of autoimmune diseases resembling human SLE. These studies indicate that negative regulation of PRRs, especially TLR signaling, is important for coordinated innate immune responses. It is well known that TLR-dependent inflammation contributes significantly to the pathogenesis of ischemia-reperfusion myocardial injury. Mice lacking TLR2, TLR4, or MyD88 show reduced infarcted regions in response to cardiac as well as cerebral ischemic-reperfusion injury (Arumugam et al., 2009). In contrast, pretreatment of mice with TLR ligands such as LPS induces tolerance and reduces the infarct size after ischemicreperfusion injury. The effects of TLR agonists may be explained by tolerance induction through initial stimulation of TLRs. TLRs are similarly involved in the pathogenesis of cerebral ischemiareperfusion injury models. Other extensive studies have shown that TLR2 and TLR4 are required for the production of atherosclerotic lesions in mice with hyperlipidemia. Given that TLR2 and TLR4 are clearly responsible for the severity of inflammation induced by nonmicrobial agents, it has been postulated that endogenous molecules directly activate these TLRs. It has been shown that HMGB1 released from necrotic cells stimulates TLR2 and TLR4 to induce the production of proinflammatory cytokines (Lotze and Tracey, 2005). Furthermore, heat shock proteins (HSPs) such as HSP60, HSP70, gp96, and HSP22 are also reported to be recognized by TLR2 and TLR4 (Asea et al., 2002; Ohashi et al., 2000; Roelofs et al., 2006; Warger et al., 2006). However, most studies describing endogenous protein ligands for TLR2 and TLR4 used recombinant proteins generated in an Escherichia coli expression system. We need to interpret the data carefully as it is very difficult to completely eliminate the possibility of contamination by E. coli components that stimulate TLRs. Nevertheless, it is apparent that these TLRs contribute to inflammatory diseases caused by nonmicrobial agents. Further studies will uncover how these agents activate TLR-induced inflammation.
'Pattern Recognition Receptors and Inflam' 카테고리의 다른 글
| Pattern Recognition Receptors and Inflammation - TLRs and Their Ligands (0) | 2024.12.04 |
|---|---|
| Pattern Recognition Receptors and Inflammation - Introduction (0) | 2024.12.03 |