Many of the most widespread and devastating diseases in humans and livestock are caused by viruses and bacteria that enter cells for replication. Being obligate intracellular parasites, viruses have no choice. They must transport their genome to the cytosol or nucleus of infected cells to multiply and generate progeny. Bacteria and eukaryotic parasites do have other options; most of them can replicate on their own.
However, some have evolved to take advantage of the protected environment in the cytosol or in cytoplasmic vacuoles of animal cells as a niche favorable for growth and multiplication. In both cases (viruses and intracellular bacteria), the outcome is often destructive for the host cell and host organism. The mortality and morbidity caused by infectious diseases worldwide provide a strong rationale for research into pathogen–host cell interactions and for pursuing the detailed mechanisms of transmission and dissemination.
The study of viruses and bacteria can, moreover, provide invaluable insights into fundamental aspects of cell biology. Here, we focus on the mechanisms by which viral and bacterial pathogens exploit the endocytosis machinery for host cell entry and replication. Among recent reviews on this topic, dedicated uniquely to either mammalian viruses or bacterial pathogens, we recommend the following: Cossart and Sansonetti (2004); Pizarro-Cerda and Cossart (2006); Kumar and Valdivia (2009); Cossart and Roy (2010); Mercer et al. (2010b); Grove and Marsh (2011); Kubo et al. (2012); Vazquez-Calvo et al. (2012a); Sun et al. (2013).
The term “endocytosis” is used herein in its widest sense, that is, to cover all processes whereby fluid, solutes, ligands, and components of the plasma membrane as well as particles (including pathogenic agents) are internalized by cells through the invagination of the plasma membrane and the scission of membrane vesicles or vacuoles.This differs from current practice in the bacterial pathogenesis field, where the term “endocytosis” is generally reserved for the internalization of molecules or small objects, whereas the uptake of bacteria into nonprofessional phagocytes is called “internalization” or “bacterial-induced phagocytosis.”
In addition, the term “phagocytosis” is reserved for internalization of bacteria by professional phagocytes (macrophages, polymorphonuclear leucocytes, dendritic cells, and amoebae), a process that generally but not always leads to the destruction of the ingested bacteria (Swanson et al. 1999; May and Machesky 2001; Henry et al. 2004; Zhang et al. 2010).
With a few exceptions, we will not discuss phagocytosis of bacteria or the endocytosis of protozoan parasites such asToxoplasma and Plasmodium (Robibaro et al. 2001).
VIRUSES
Viruses are lifeless particles lacking metabolism and means of locomotion. Their mission (raison d’eˆtre) is to serve as carriers for their own genome during cell-to-cell spread and organism-to-organism transmission. They protect the genome (RNA or DNA) during transit, and deliver it into the cytosol or the nucleus of new host cells usually together with accessory proteins.
Although often spherical, mammalian viruses can be bullet-shaped, brick-shaped, amorphous, or filamentous ranging in diameter from 18 to 2000 nm. In enveloped animal viruses, the membrane is a de facto transport vesicle. It is formed by budding (fission) from membranes in the infected cell. It releases its cargo (a nucleocapsid and accessory proteins) by fusing with a membrane in the new host cell.
In this way, there is no need for macromolecule complexes to ever cross a membrane. To overcome the membrane barriers, nonenveloped viruses induce membrane lysis, generate pores, or rely on membranecrossing devices provided by the cell. The majority of nonenveloped and enveloped viruses depend on endocytosis for entry. This means that the penetration reaction—whether by fusion or other mechanisms—occurs in intracellular organelles and involves intracellular membranes of the host cell.
BACTERIA
Although the simplest among living organisms, bacteria are considerably more complex than viruses. They are single-celled and have different shapes (spherical, spiral, or rod shaped) and appear singly or in chains. A typical bacterium is 1–5 mm in length. It has a cell membrane and a rigid cell wall, and it lacks a nucleus. Some have an additional outer membrane, and some have appendages such as flagella that they use to move in the environment or pili that they use for adhesion to inert surfaces, host cells, and even to other bacteria. In contrast to viruses, bacteria can secrete proteins including toxins via a variety of secretion systems, classified from I to VII depending on the structure and organization of the secretion machinery.
Highly relevant for this review are the type III and the type IV secretion systems, which appear as nanomachines on the bacterial surface (Backert and Meyer 2006; Thanassi et al. 2012). They are dedicated to the transport of effector proteins directly from the interior of the bacteria into the interior of the host eukaryotic cell. These effector proteins are either enzymes that use host components as substrates or proteins that structurally or functionally mimic eukaryotic proteins and thus interfere with cellular mechanisms (Galan and Wolf-Watz 2006; Agbor and McCormick 2011).
Many pathogenic bacteria multiply extracellularly, but it is increasingly recognized that many bacteria that were long considered extracellular can also reside, replicate, or persist inside cells (Pizarro-Cerda and Cossart 2006). Depending on the type of host cells (phagocytic or nonphagocytic) with which bacteria interact, there are two possible scenarios. In phagocytic cells such as macrophages, cells are the active players in the internalization process; they engulf bacteria and internalize them. In contrast, in nonphagocytic cells, only a few bacterial species—qualified as “invasive”—enter as the result of a process that the bacteria initiate.
Some bacteria have obligate intracellular life styles like viruses; that is, they cannot replicate outside a eukaryotic cell. Generally, these bacteria have lost genes required for independent replication in broth medium and have small genomes. Nevertheless, as recently shown for Coxiella burnetii, the use of complex media may provide nutrients for extracellular replication of pathogens up to now considered as obligate intracellular pathogens (Omsland et al. 2009).
ENDOCYTOSIS OF VIRUSES AND BACTERIA: THE GENERAL PICTURE
There are many reasons why incoming pathogens enter host cells by endocytosis for replication. In the case of bacteria, entry into cells and replication therein are believed to protect them from circulating antibodies and complementinduced destruction. Yet, inside cells, pathogens have to confront a variety of cellular defense mechanisms. They have evolved numerous sophisticated mechanisms to counteract these bactericidal processes. Specifically, they may secrete proteins or components that either allow modification of the internalization vacuole to permit an intravacuolar lifestyle with concomitant replication or trigger escape from the vacuole (Kumar and Valdivia 2009).
Of the bacteria that escape from vacuoles, some are able to recruit actin and move intra- and intercellularly (Gouin et al. 2005). Others simply multiply without further movement within the nutrient-rich environment of the cytosol. In this case, cell lysis leads to pathogen dissemination. It is to be noted that for intracytosolic bacteria, entry into the cytosol involves lysis of intracellular vacuoles. This is likely to be less damaging for the cell than if it occurred directly through the plasma membrane.
For viruses, endocytic vesicles help to ferry the incoming particles deep into the cytoplasm unobstructed by cytoplasmic crowding and obstacles such as the cytoskeleton. In the process of intracellular maturation of endocytic vacuoles, the host cell exposes the viruses to changing conditions including a drop in pH that many viruses use as a cue to activate penetration (Helenius et al. 1980; Vazquez-Calvo et al. 2012a).
Exposure to proteases and processing of viral proteins is critical for some viruses because it allows activation of viral penetration and uncoating mechanisms (Danthi et al. 2010; Hunt et al. 2012; Kubo et al. 2012). By fusing their envelope with membranes of internal organelles, enveloped viruses can, moreover, avoid exposing their glycoproteins on the cell surface, and can thus presumably delay detection by immune surveillance. Some enveloped viruses belonging to the retro-, paramyxo-, pox-, and herpesviruses can, however, release their capsids into the cytosol by fusing their envelope membrane with the plasma membrane.
In most cases, it is not clear whether such fusion results in infection because virus particles are also being endocytosed in the same cells, and these could be the ones causing infection.
Unlike many of the invasive bacteria, incoming viruses cannot manipulate the endocytic machinery by prior delivery of effector proteins into the cytosol of host cells. To induce endocytosis and prepare the host cell for invasion, they make use of the cell’s signaling and regulatory pathways, but they do so indirectly from the plasma membrane.
VIRUS BINDING TO HOST CELLS
Endocytic entry of viruses occurs in a stepwise manner involving attachment to the cell surface, clustering of receptors, activation of signaling pathways, formation of endocytic vesicles and vacuoles, delivery of viral cargo to endosomal compartments, sorting, and escape into the cytosol. Depending on the virus, capsid escape occurs from early endosomes, late endosomes, lysosomes, macropinosomes, or the endoplasmic reticulum (ER) (Fig. 1; Table 1).

Figure 1. Viruses in the endocytic network. After binding to cell-surface receptors, viruses are internalized through a variety of endocytic processes including macropinocytosis, clathrin-mediated endocytosis, caveolae, and clathrin- and caveolin-independent mechanisms. Some of the viruses that use these mechanisms of endocytosis are listed. The primary endocytic vesicles and vacuoles formed ferry incoming virus particles into the endocytic network, where they undergo sorting and eventually penetration into the cytosol from different locations within the vacuolar network. Five different locations are indicated by the numbered gray arrows. Pathways followed by influenza A virus, SV40, Uukuniemi virus, and vaccinia virus are shown. Of these, SV40 is transported to the endoplasmic reticulum for uncoating and penetration, whereas influenza A and vaccinia virus undergo acid-activated membrane fusion in maturing and late endosomes and macropinosomes, respectively. The location and timing of escape are often determined by the pH threshold for penetration of the virus particles as indicated. LCMV, lymphocytic choriomeningitis virus.


Possible additional locations include the trans-Golgi network (TGN), the Golgi complex, recycling endosomes, or amphisomes (Suikkanen et al. 2002; Berryman et al. 2012; Day et al. 2013; Lipovsky et al. 2013).
Viruses only infect cells to which they can bind. The choice of receptors and the specificity of binding contribute to the choice of entry mechanism, to species and tissue specificity of infection, to cell tropism, and ultimately to the course of the disease. Enveloped viruses bind via spike glycoproteins, whereas nonenveloped viruses usually attach via fibers, spikes, or surface indentations. Ranging from proteoglycans to glycolipids and glycoproteins, the overall spectrum of cellular molecules identified as virus receptors is extremely broad (Helenius 2007).
Some serve merely for attachment and concentration of viruses to the cell surface, whereas others have additional roles in signaling, endocytosis, and in the modification of the bound virus. In many cases, virus–cell interactions involve carbohydrates (Vasta 2009; Kamhi et al. 2013). Although well-known endocytic receptors such as transferrin and LDL receptors occur in the list, the majority of virus receptors are not directly involved in the uptake of physiological ligands. By inducing clustering, viruses can generate a receptor-rich membrane microdomain that differs from the rest of the plasma membrane in protein and lipid composition (English and Hammer 2005; Lozach et al. 2011b). Such domains may serve as a platform for transmembrane signaling, for recruitment of cytoplasmic coats, and for assembly of endocytic machinery. In some instances, antibodies, complement factors, and other proteins in body fluids can play a bridging function between the virus and its receptors (Flipse et al. 2013).
Receptors usually follow the virus into endocytic vesicles and into the cell. In addition to the virus particle, the endocytic “cargo” thus comprises a complex of receptors and lipids. Importantly, virus–receptor interactions can result in alterations in the structure of viral proteins or in the virus particle as a whole (Lonberg-Holm and Philipson 1974; Raff et al. 2013). Simultaneous binding to a mobile and an immobile receptor was, for example, recently shown to subject adenovirus particles to forces that initiate uncoating by detaching some of the viral fibers (Burckhardt et al. 2011). A receptorinduced conformational change in the glycoprotein of HIV-1 is essential for triggering penetration by membrane fusion (Wilen et al. 2012). In recent years, a new class of interaction partners on the cell surface has been recognized for viruses. These could be called “accessory factors” because they are primarily needed to activate signaling pathways and thus trigger endocytosis. They include receptor tyrosine kinases, phosphatidylserine (PS) receptors (TIM and TAM receptors), and integrins (Mercer and Helenius 2010; Morizono et al. 2011; Meertens et al. 2012).
The interaction of viruses with these can be mediated by adaptors such as Gas6, a soluble protein that bridges between PS in the viral membrane and receptor tyrosine kinase Axl during activation of virus uptake by macropinocytosis (Morizono et al. 2011; Meertens et al. 2012). The physiological role of Gas6 is to serve as a soluble adaptor protein between PS in the membrane of cell remnants after apoptosis and Axl on the surface of cells (Lemke and Burstyn-Cohen 2010). It is often observed that viruses use more than one type of receptor either in parallel, in sequence, or when interacting with different cell types. Rotaviruses, hepatitis C virus, HIV-1, and coxackie B virus are examples of viruses that interact sequentially with different cell-surface molecules (Coyne and Bergelson 2006; Lopez and Arias 2006). Other viruses such as many herpesviruses carry multiple receptor-binding glycoproteins with different specificity and can therefore enter different cell types (Eisenberg et al. 2012). That viruses can exploit multiple endocytic mechanisms and pathways is also clear. Herpes simplex virus I uses different pathways in different cell types (Campadelli-Fiume et al. 2012). Influenza Aviruses can, for example, use two or three different pathways (Matlin et al. 1981; Sieczkarski and Whittaker 2002; Lakadamyali et al. 2006; de Vries et al. 2011). Considering the underlying redundancy and complexity in virus–cell interactions, it is not surprising that the literature on the entry of many viruses is inconsistent and confusing.
BACTERIAL BINDING TO HOST CELLS
Endocytosis of bacteria starts by an irreversible interaction. Two main mechanisms may take place. In the first type, the adhesion between a bacterial ligand and a specific host surface receptor causes receptor clustering, the zippering of the plasma membrane around the bacterium, and the initiation of signaling events that culminate in entry. It is often difficult to separate the steps of adhesion and endocytosis during entry by this “zipper mechanism.”
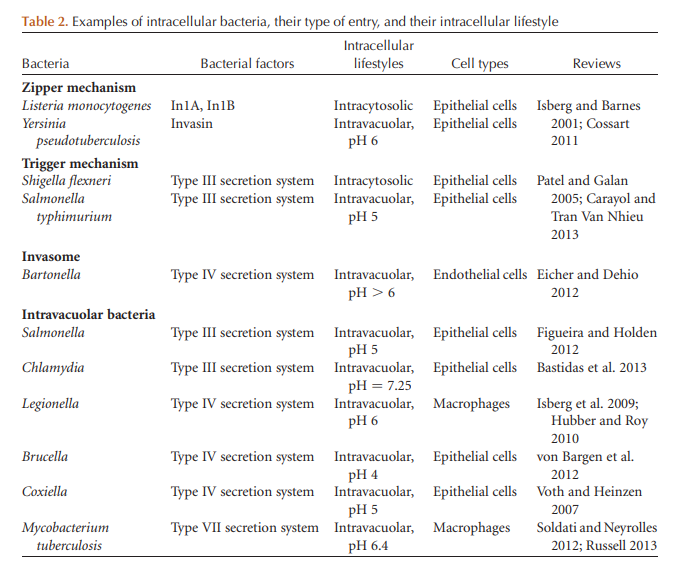
In the second mechanism of bacterial endocytosis, the initial contact is a relatively transient event that essentially permits the direct delivery into the cell cytosol of active T3SS effectors that trigger cytoskeleton rearrangements and membrane remodeling, which lead to a macropinocytosislike event. This second way of entry is called “the trigger mechanism” (Fig 2; Table 2) (Cossart and Sansonetti 2004).

Figure 2. Pathways of endocytic entry by invasive bacteria. Schematic representation of bacterial endocytosis mechanisms and intravacuolar or intracytosolic lifestyles. In epithelial cells, Listeria enters via two internalins; it then resides transiently in a vacuole that is lysed allowing intracytosolic replication and actin-based motility. The obligate intracellular bacterium Chlamydia trachomatis resides in a vacuole that intercepts the pathway involved in the transport of shingomyelin from the Golgi apparatus to the plasma membrane. Shigella is first taken up by filopodia present on the surface of epithelial cells. The type 3SS then injects effectors that trigger entry, formation of the vacuole, and escape from this compartment, leading to intracytosolic replication and actin-based motility. Salmonella appears to enter after a “near-surface swimming” mechanism. The T3SS-1 (or SPI-1) triggers entry and formation of the replicative vacuole, which acquires markers of endosomes and lysosomes owing to the secretion of effectors of the T3SS-2 (or SPI-2). In some circumstances, Salmonella can reach the cytosol and rapidly replicate therein. The Brucella replicative vacuole is derived from the endocytic vacuole and matures in an ER-derived vacuole. Coxiella burnetii is the only bacterium that has evolved to survive and replicate in a lysosome-derived vacuole. Bartonella henselae can enter into endothelial cells as a single bacterium or as a group, leading to the formation of an invasome. In macrophages, Legionella and Mycobacterium tuberculosis reside in a vacuole. The Legionella vacuole acquires markers of the ER. It is not the case for M. tuberculosis, which blocks the maturation of the internalization vacuole.
Binding to cells occurs via proteins called “adhesins” located either on the bacterial surface or at the tip of appendages called “pili” or “fimbriae,” which form hair-like fibers that protrude from the surface. Because pili can be used for the transfer of genetic material during conjugation, the term “fimbriae” has in the past been used to describe those pili whose specific function is adhesion. However, the nomenclature remains fuzzy.
Interestingly, some pili (e.g., type IV pili) not only mediate bacteria–host cell or bacteria–bacteria adhesion but also perform complex functions such as force-driven contraction providing bacteria with powerful tools to enhance their contact with target surfaces.
One of the best-characterized fimbriae is the pyelonephritis-associated (P) pilus, expressed by uropathogenic Escherichia coli that colonize the urinary tract and then infect the kidney. P pili bind through the PapG adhesin, located at the tip of the complex pilus rod, to the aD-galactopyranosyl(1-4)-b-D-galactopyranoside moiety of glycolipids of upper urinary tract cells (Waksman and Hultgren 2009).
In addition, the pili mediate the generation of biofilm-like structures on and inside host cells, contributing to the persistence of the bacteria in the bladder. Type IV pili are essentially composed of a homopolymer of a single pilin subunit such as pilA in Pseudomonas aeruginosa, PilE in Neisseria spp., or TcpA in Vibrio cholerae (Craig et al. 2004). As stated above, type IV pili have been implicated in other functions including biofilm formation. Interestingly, a posttranslational modification of the Neisseria meningitidis pilin subunit has been shown to change the conformation of the pilin and lead to loss of interbacterial interactions at sites of adhesion leading to Neisseria dissemination through epithelial cells and bacterial access to blood vessels (ChamotRooke et al. 2011).
Recently a novel type of pilus made of a single type of pilin subunit—polymerized via sortases that covalently link the subunits together—was identified in clinically important pathogens such as Streptococci and Pneumococci and shown to be critical for infection paving the way to new vaccine strategies (Ton-That et al. 2004; Dramsi et al. 2006; Falker et al. 2008). A variety of nonpolymeric adhesins have been reported that mediate bacterial adhesion to host surface components, in particular, to host membrane adhesion receptors such as integrins, cadherins, selectins, and CEACAMs. In the case of invasive bacteria, these adhesins often serve as invasion proteins, that is, proteins that mediate the endocytosis process itself (Fig. 2).
It is the case for Internalin (InlA) of Listeria monocytogenes (Gaillard et al. 1991) or invasin of Yersinia pseudotuberculosis (Isberg et al. 1987) (see below). A unique situation has been described in the case of enteropathogenic E. coli. These bacteria inject a type III effector called Tir into mammalian cells. Tir inserts in the plasma membrane and acts as a receptor for the bacterial protein intimin (Rosenshine et al. 1996; Kenny et al. 1997). Tir also mediates the recruitment of several proteins including clathrin, dynamin, and Dab2 before the recruitment of components of the actin cytoskeleton and, in particular, the Arp2/3 complex, which mediates the polymerization of actin filaments (Unsworth et al. 2007; Veiga et al. 2007; Bonazzi et al. 2011). The result is the formation of a pedestal on which bacteria are located and tightly adhere. Hence, tight adherence in the presence of typical endocytic molecules such as clathrin or dynamin does not necessarily lead to endocytosis.
ENDOCYTOSIS OF VIRUSES
The rate and efficiency of viral endocytosis are variable. After binding to the cell surface, internalization can occur with a half-time as short as a few minutes or as long as several hours. Uptake is usually nonsynchronous, but in the end quite efficient. Viruses exploit the capacities of the host cell by making use of different cellular endocytosis mechanisms, which they can activate and modify for their specific purposes when needed (Fig. 1). Although the dependence of viruses on cellular processes in all stages of entry and uncoating makes possible the stripped-down economy in the structure of viral particles, it also means that viruses and their components must interact directly or indirectly with a multitude of cellular factors.
Clathrin-Mediated Endocytosis
Clathrin-mediated endocytosis (CME) is probably the most common mechanism for endocytosis of small and medium-size viruses (Figs. 1 and 3; Table 1) (Dales 1978; Helenius et al. 1980; Matlin et al. 1981; DeTulleo and Kirchhausen 1998; Sun et al. 2013).
Time-lapse movies show two general behaviors. After a period of lateral movement along the membrane, some particles diffuse into preexisting clathrin-coated areas, and others induce clathrin assembly at the site of binding (Ehrlich et al. 2004; Rust et al. 2004; van der Schaar et al. 2008; Johannsdottir et al. 2009). In the latter case, the assembly of a clathrin coat takes somewhat longer than for clathrin pit formation during uptake of endogenous protein ligands (1–6 min). The coated vesicles pinch off with one or more viruses. The coat dissociates within 5–20 sec, and the viruses are delivered within minutes to early endosomes in the periphery of the cell.
Like CME of physiological cargo, uptake of viruses depends on PI(4,5)P2 and dynamin-2 and is usually inhibited by chlorpromazine, an inhibitor of CME (Wang et al. 1993; Sieczkarski and Whittaker 2002). Vesicular stomatitis virus (VSV) belongs to the viruses that use CME for productive infection (Matlin et al. 1982). In human cells, the receptor for VSV was recently identified as the LDL receptor and its family members (Finkelshtein et al. 2013). Light microscopy in live cells combined with single-particle tracking shows that the clathrin coat is recruited to sites of VSV binding (Cureton et al. 2009; Johannsdottir et al. 2009). Because of the bullet shape and large size of VSV particles, the clathrin vesicles form slowly, they deviate from the round shape, and some carry only a partial clathrin coat (Cureton et al. 2009). Although VSV endocytosis is dependent on plasma membrane PI(4,5)P2, dynamin, and actin, it is unclear whether AP2 is the adaptor protein required for endocytosis and infection (Cureton et al. 2009; Johannsdottir et al. 2009; Vazquez-Calvo et al. 2012b). A general lesson from the observations with VSV and other viruses is that although the viruses differ in size, they have evolved to use CME very efficiently. They can induce the formation of clathrin-coated vesicles (CCVs) locally, and they can adapt the size and shape of the clathrincoated pits (CCPs) for their needs. It will be interesting to determine how VSV and other viruses guide and regulate the coat assembly process.
Clathrin- and Caveolin-Independent Endocytosis
The existence of a clathrin-, caveolin-, and dynamin-independent pathway for virus endocytosis was first described for members of the polyomaviruses: mouse polyoma virus, and SV40 (Gilbert and Benjamin 2000; Damm et al. 2005). These are small nonenveloped DNA viruses (diameter 50 nm) that replicate in the nucleus of host cells. The VP1 proteins that form the icosahedral viral coat bind to the sialic-acid-containing carbohydrate moiety of specific gangliosides that serve as cell-surface receptors (Tsai et al. 2003). Present as 72 homopentamers, VP1 constitutes the main building block in the capsid shell (Stehle et al. 1996). By binding to multiple GM1 molecules, SV40 actively generates membrane curvature (Ewers et al. 2010). In a process that in EM sections looks as if the virus particle would be budding into the cell, the PM wraps itself tightly around the virus particle (Fig. 4F). The same can be seen when viruses are added to giant unilamellar liposomes containing GM1 with long acyl chains. When many viruses are present, long, narrow, tight-fitting, tubular invaginations with multiple viruses can form in the PM of cells and in liposomes. Interestingly, pit and tube formation does not require the whole virus; isolated VP1 homopentamers are sufficient. For the fission reaction that detaches the virus-containing indentations, the virus alone is not sufficient. Energy in the form ATP is required, as well as cellular tyrosine kinases, cholesterol, and a change in actin dynamics. Contrary to CCVs, vesicle fission is independent of dynamin (Damm et al. 2005). The viruscontaining vesicles formed are delivered to early endosomes, and the virus is eventually transported via late endosomes to the ER (Fig. 1) (Qian et al. 2009; Engel et al. 2011). It is noteworthy that a similar mechanism is used by pentameric, bacterial toxins (cholera and Shiga) that bind to glycolipids (Ro¨mer et al. 2010). In all these cases, the receptors are glycosphingolipid molecules concentrated in lipid rafts. They differ from most other receptors in that they do not span the bilayer.
Caveolar Endocytosis
Caveolar endocytosis has been proposed for a variety of viruses. One of the problems in assigning viruses to this pathway is that the endocytic role of caveolae in cell life is still poorly defined. No single characteristic such as cholesterol dependence alone is sufficient as a criterion to define this pathway. Although much less dynamic than CCVs, it is clear, however, that a fraction of the caveolae is mobile and can undergo endocytosis especially when activated by a ligand (Kirkham and Parton 2005; Pelkmans and Zerial 2005; Tagawa et al. 2005). The best studied among the viral candidates for caveolar uptake is SV40. Morphological evidence from several laboratories using immune-electron and immunofluorescence microscopy shows colocalization of the viruses and caveolin-1 in plasma membrane spots and pits (Anderson et al. 1996; Stang et al. 1997; Pelkmans et al. 2001). Addition of SV40 to cells causes a dramatic, tyrosine-phosphorylationdependent elevation in caveolar vesicle formation and caveolar vesicle motility (Tagawa et al. 2005). shRNA- and siRNA-mediated depletion of caveolin-1 results in a decrease in SV40 endocytosis and infection (Pelkmans et al. 2004; Stergiou et al. 2013). Recently, it was shown that depletion of EHD2 (a peripheral caveolar ATPase) leads to a dramatic increase in caveolar vesicle trafficking and increases the efficiency of SV40 infection (Stoeber et al. 2012; M Stoeber, G Balustreri, and A Helenius, unpubl.). Thus, in summary, it seems that SV40 can make use of two parallel endocytic mechanisms. One corresponds to the caveolar pathway and relies on caveolin-1. The other is caveolin independent, with the virus particle serving as the curvature-generating principle. To what extent viruses other than SV40 can use these pathways is not clear. There are reports suggesting that other polyomaviruses such as BK virus and mouse polyoma virus may also exploit caveolae (Richterova et al. 2001; Neu et al. 2009).
Virus-Triggered Macropinocytosis
Macropinocytosis offers incoming viruses an altogether different type of endocytic experience (Fig. 1). Transiently triggered by external ligands such as growth factors and PS-containing particles, this mechanism is known to induce internalization of fluid, membrane, and whatever happens to be associated with the membrane such as viruses (Swanson and Watts 1995). The process is important for the clearing of cell remnants after apoptotic cell death. The PS exposed on cell remnants constitutes an “eatme” signal that secures efficient uptake and degradation of cellular garbage without triggering an inflammatory response (Hoffmann et al. 2001). The key events in macropinocytosis include the activation of receptor molecules such as receptor tyrosine kinases, integrins, and PS receptors (Swanson and Watts 1995). This triggers downstream signaling cascades resulting in transient, global changes in actin dynamics that lead to cell-wide plasma membrane ruffling in the form of filopodia, lamellopodia, circular ruffles, or blebs. Large, uncoated vacuoles (macropinosomes) are formed. After moving deeper into the cytoplasm, these usually end up fusing with late endosomes or lysosomes. Important distinguishing features for macropinocytosis include activation Rho GTPases (Rac1 and/or Cdc42), myosins, and kinases such as PAK1, PI3 kinase, and PKC. The process is sensitive to inhibitors of Naþ/Hþ exchangers. Because there are many variations of the process, macropinocytosis must be viewed as a collective term for an assortment of related mechanisms. Macropinocytosis plays a role in the infection of viruses of different families including large viruses such as pox-, filo-, paramyxo, and herpesviruses (Lim and Gleeson 2011; Mercer and Helenius 2012). Some smaller enveloped and nonenveloped viruses such as influenza A virus seem to use pathways that share many properties with macropinocytosis (Khan et al. 2010; de Vries et al. 2011). By exposing PS in their envelope, vaccinia and some other enveloped viruses make use of “apoptotic mimicry” (Mercer and Helenius 2008). The emerging role of PS receptors of the TIM and TAM families and adaptors such as Gas6 in this process was already discussed above. Epidermal growth factor receptor (EGFR) and other growth factor receptors are often activated and essential as accessory factors in infectivity (Eierhoff et al. 2010; Mercer et al. 2010a; Krzyzaniak et al. 2013).
Other Endocytic Pathways for Virus Internalization
There are viruses for which endocytic entry does not fall into the categories listed above (Table 1). One of them is human papilloma virus 16 (HPV-16), which in HeLa and HaCaT cells uses a mechanism with similarities to macropinocytosis (Schelhaas et al. 2008). However, the vesicles are small, Rho GTPases are not activated, and there is no elevation in fluid uptake. Some of these characteristics are shared by human rhinovirus 14 and one of the alternate pathways described for influenza A virus (Matlin et al. 1981; Rust et al. 2004; Khan et al. 2010; de Vries et al. 2011). In addition, two Old World arenaviruses, LCMV and Lassa virus, enter by a clathrin-, caveolin-, and dynamin-independent pathway via multivesicular bodies bypassing early endosomes (Quirin et al. 2008; Kunz 2009). In the case of Acanthamoeba polyphaga mimivirus, a giant enveloped virus (750 nm in diameter) that uses amoeba as a host, uptake into human macrophages seems to involve a phagocytosis-like mechanism (Ghigo et al. 2008). This uptake process resembles the phagocytosis of bacteria. Of the various endocytic mechanisms described in mammalian cells, the CLIC/GEEC, IL2, flotillin, and CIE/Arf 6 pathways do not seem to support virus entry (Lamaze et al. 2001; Mayor and Pagano 2007; Donaldson et al. 2009; see also Mayor et al. 2014). However, because virus entry studies and the classification of endocytic mechanisms are not always straightforward, the situation may change. Viruses may, in fact, provide one of the most informative systems for further classification of endocytic processes.
ENDOCYTOSIS OF BACTERIA
The rate and efficiency of entry of bacteria into cells vary greatly with the cell type and other parameters such as the temperature or the growth phase at which bacteria have been harvested (Fig. 5).

Figure 5. Endocytosis of bacteria. These electron micrographs show various phases of bacterial endocytosis into mammalian cells. (A, left) Binding of Listeria entering into cells. Two coated pits are detectable on this cross section. The tight apposition of the membrane illustrates what is meant by the zipper mechanism. (A, middle micrograph) A Shigella entering into a cell and the huge membrane ruffles that engulf the bacterium. This micrograph illustrates the trigger mechanism. (A, right) Bartonella henselae enter as a group into an endothelial cell. (B) Listeria is entering into the cell and the clathrin coat is visible as a thickening of the plasma membrane underneath the bacterium. (C, left) Listeria is present in a membrane-bound vacuole. (C, right) Chlamydia vacuole full of bacteria that have replicated.
The Zipper Mechanism: AClathrin- and ActinMediated Internalization Process
The hallmarks of the zipper mechanism are a dedicated bacterial surface protein or component that interacts directly with a host cell receptor, thereby inducing a series of signaling events that culminates in endocytosis. The actin cytoskeleton and its dynamics as well as the membrane composition and its plasticity are critical elements (Pizarro-Cerda and Cossart 2004, 2006; Cossart and Roy 2010). The bacterial proteins involved in entry often mimic endogenous ligands and exploit the properties of their receptors maximally. Alternatively, bacteria can (like viruses described above) interact with components that act as a bridge to a cellsurface receptor. It is clear that the affinity of the ligand and its density on the bacterial surface critically control the efficiency of the entry process. More recently, the CME machinery has been shown to be involved in early steps after initial contact between the incoming microbe and the receptor before the cytoskeleton rearrangements (Boleti et al. 1999; Veiga and Cossart 2005; Veiga et al. 2007; Eto et al. 2008; Pizarro-Cerda et al. 2010; Bonazzi et al. 2011). Listeria monocytogenes is the prototype of a bacterium entering by the zipper mechanism (Pizarro-Cerda et al. 2012). It expresses two proteins involved in entry. The first, Internalin (InlA), is a surface protein that interacts with E-cadherin, a cell–cell adhesion molecule expressed only in some epithelial cells (Mengaud et al. 1996). For entry into most cell types, Listeria uses a second invasin, the InlB protein, which activates the receptor tyrosine kinase Met, the hepatocyte growth factor receptor that is expressed on all cells of epithelial origin (Shen et al. 2000; Bierne and Cossart 2002). Integrity of the membrane and its lipid rafts is critical for the initial clustering of E-cadherin when bacteria enter by the InlA pathway. In contrast, for the InlB pathway, the integrity of lipid rafts is critical for the correct localization of the phosphoinositides produced in the plasma membrane by PI3 kinase, which is recruited to the receptor activated upon bacterial entry (Seveau et al. 2004, 2007). Investigations of Listeria endocytosis in cells that do not express E-cadherin have shown that InlB mimics HGF and first induces the autophosphorylation of Met (Shen et al. 2000). This, in turn, leads to the recruitment of Gab1, Cbl, and Shc (Ireton et al. 1999; Shen et al. 2000). The ubiquitin ligase Cbl triggers the ubiquitination of Met as a prelude for the recruitment of the clathrin adaptor Dab2 and that of the clathrin heavy and light chains (Veiga and Cossart 2005). Src-mediated phosphorylation of the clathrin heavy chain is critical for the whole process (Veiga et al. 2007; Bonazzi et al. 2011). Strikingly, Hip1R, which interacts with the clathrin light chain and actin filaments, is then recruited followed by myosin VI, which interacts with Hip1R and actin. Because myosin VI has the capacity to move toward the minus end of actin filaments, it probably then pulls the bacteria toward the interior of the cell. Dynamin is also recruited at the bacterial entry site, as well as cortactin. A first wave of actin polymerization may thus take place following the interaction of cortactin with the Arp2/3 complex. A second wave of actin rearrangements is then triggered more classically by the Gab1-mediated recruitment of PI3 kinase (Ireton et al. 1996) and the activation of small G proteins (Pizarro-Cerda et al. 2012). Note that actin rearrangements are finally down-regulated by the recruitment of proteins such as cofilin (Bierne et al. 2001) or OCRL a phosphatidyl inositol-5-phosphatase that dephosphorylates PI(4,5)P2 and PI(3,4,5) P3 (Kuhbacher et al. 2012). Strikingly, although immunofluorescence data clearly show clathrin recruitment at the entry site (Veiga and Cossart 2005; Veiga et al. 2007), ultrastructural analysis by electron microscopy of cells infected with Listeria shows the presence of clathrin on membranous invaginations, as isolated clathrin-coated pits (Bonazzi et al. 2011). Clathrin-coated vesicles are not seen. That clathrin depletion prevents actin recruitment suggests that coated pits serve as a platform for cytoskeletal arrangements (Lecuit et al. 2000; Pizarro-Cerda et al. 2010; see also Brodsky et al. 2014). In cells that express E-cadherin, Listeria exploits the normal properties of this transmembrane protein and induces its interaction with a and b catenins to trigger signaling events that culminate in Arp2/3-dependent actin polymerization events (Sousa et al. 2007). Interestingly like Met (see above) and many other receptors, E-cadherin undergoes several posttranslational modifications upon activation including phosphorylation and ubiquitination (Bonazzi et al. 2008). Ubiquitination is mediated by the E-cadherin-specific ubiquitin ligase Hakai and allows the recruitment of clathrin and other components of the clathrin-mediated endocytosis machinery upstream of actin rearrangements (Bonazzi et al. 2008). Interestingly, colocalization of caveolin at the site of the InlA E-cadherin entry site and depletion experiments show that caveolin also participates in entry. Listeria entry is thus a complex process implicating the clathrin-mediated endocytosis machinery, the actin cytoskeleton as in classical phagocytosis, and caveolae (Pizarro-Cerda et al. 2012). Entry of Yersinia pseudotuberculosis into cells resembles that of Listeria (Wong and Isberg 2005). An outer membrane protein called invasin interacts with b1-integrins, that is, proteins that are normally implicated in adherence of cells to the extracellular matrix (Van Nhieu and Isberg 1991). Unlike fibronectin, invasin does not possess an RGD motif, but has a domain structurally similar. Invasin has a higher affinity than fibronectin for integrins. It induces integrin clustering and efficient downstream signaling. The cytoplasmic tail of the b1 chain, which normally interacts with the cytoskeleton in focal complexes of adhesion plaques, is critical for entry, but, surprisingly, alterations in this domain that impair interaction with the cytoskeleton increase internalization (Van Nhieu et al. 1996). Thus, a lower affinity of the integrin for the cytoskeleton could allow higher mobility of the receptors in the membrane. Activation of integrins leads to the activation of the Arp2/3 complex and to the actin rearrangements necessary for uptake. The local concentration of PI(4,5)P2 is critical for entry, and Arf6 may have a role in activation of PIP5 kinase, the control of cytoskeleton rearrangements and membrane traffic involved in the closure of the phagocytic cup (Wong and Isberg 2003). The analysis of Yersinia entry has frequently involved the use of E. coli-expressing invasin as a surrogate. This strain has recently been used to show that (as for Listeria) the clathrin-mediated endocytosis machinery is critical for bacterial en try (Veiga et al. 2007). However, this has not been studied in detail yet. Several other bacteria such as some Streptococci and also Staphylococcus aureus use integrins for their uptake (Ozeri et al. 2001; Kreikemeyer et al. 2004). However, they express a variety of fibronectin-binding proteins and use a bridging mechanism. As for Listeria, uptake of these bacteria requires components of the clathrin-mediated endocytic pathway. To enter cells, the Gram-negative bacterium Chlamydia makes use of at least one early-secreted type III secretion system (T3SS) effector called TARP, a protein that nucleates actin polymerization directly (Jewett et al. 2010). In addition to TARP, entry requires the concerted activation of growth factor receptors, cytoplasmic kinases, and small GTPases to remodel the actin cytoskeleton (Lane et al. 2008). Additional host factors include clathrin and cholesterolrich microdomains (Boleti et al. 1999; Gabel et al. 2004).
The Trigger Mechanism: A MacropinocytosisRelated Process
Shigella and Salmonella are two bacteria that use the trigger mechanism to enter cells (Tran Van Nhieu et al. 2000; Patel and Galan 2005; Carayol and Tran Van Nhieu 2013). The hallmark of this mechanism is the formation of huge, actin-rich membrane ruffles triggered by a signaling cascade that induces localized transient changes in actin dynamics. It is triggered by the translocation of T3SS effectors into the host cytosol. In Salmonella, the secretion system involved in entry is encoded by a chromosomal pathogenicity island (PAI). In Shigella, the PAI is carried by a plasmid. These PAIs encode the structural components for the T3SS and some of their dedicated effectors. For many years, the nature of the initial contact between Shigella and cells remained elusive. More recently, it was shown that upon challenge with epithelial cells, Shigella establish contacts with filopodial-like extensions, which then retract to bring bacteria into contact with the cell body, where invasion occurs (Romero et al. 2011). Filopodia are cell-surface sensory organelles implicated in adhesive processes, including the formation of intercellular junctions. They are not induced by bacterial contact. Time-lapse video microscopy showed that bacterial capture by filopodia can be inhibited by antibodies against IpaB and IpaD, two proteins that are located at the tip of the T3SS, indicating that contact likely occurs between a cell-surface receptor present at the filopodial tip and the T3SS tip complex (Carayol and Tran Van Nhieu 2013). Upon cell contact, IpaB and IpaC insert into the host cell plasma membrane to form a translocon complex that allows injection of T3SS effectors. These induce localized membrane ruffling by polymerization of cortical actin. Invasion requires the concerted action of several T3SS effectors to activate tyrosine kinases and Rho GTPases. At least two of the effectors (IpgB1 and B2) act as GEFs for Cdc42, Rac, and Rho, respectively (Bulgin et al. 2010). It is possible that these activities synergize for efficient actin polymerization. In addition to IpaC, IpgB1, and IpgB2, the IpaA and IpgD type III effectors have been implicated in Shigella invasion. IpgD may promote invasion through its PI(4,5)P2 phosphatase activity (Niebuhr et al. 2002), which might help to loosen the connection between cortical actin and the membrane, thereby affecting actin dynamics at bacterial invasion sites. IpaA induces actin depolymerization at subsequent stages of the entry process. Filopodial capture might allow bacteria to target specific sites of the epithelium (Romero et al. 2011). Indeed, Shigella invasion occurs mainly at multicellular junctions in the apical surface of polarized intestinal cells. These correspond to the intersection between several cells. Interestingly, tricellulin (a protein required for the integrity of multicellular junctions) is essential for cell-to-cell spreading of Shigella, a process involving components of the clathrin-mediated endocytosis pathway (Fukumatsu et al. 2012). In contrast to Shigella, which is nonadhesive and nonmotile and sampled by the finger-like extensions of the cell surface, Salmonella is a motile bacterium. It can, moreover, adhere to the cell via the SPI-1 T3SS (Salmonella has a second T3SS involved in intracellular replication; see below) and via the fimbrial adhesin Fim. In HeLa cells, the binding by Fim is reversible, whereas that mediated by the SPI-1 T3SS is irreversible (Misselwitz et al. 2011, 2012). It has been proposed that on a cell surface, “near-surface swimming” of Salmonella mediated by the flagellar motility allows landing on a surface, followed by Fim-mediated reversible binding with further “near-surface swimming” or docking mediated by the SPI-1 T3SS. This step commits Salmonella to invasion via the translocation of the effectors SopE, SopE2 (two GEFS for Rac and CDC42), SopB [a T3SS effector similar to IpgD with PI(4,5)P2 phosphatase activity that activates RhoG], SipC (which can nucleate and bundle actin filaments directly), and SipA (which also participates in actin polymerization) (Cain et al. 2008; Humphreys et al. 2012). The Arp2/3 complex activation via SopE/E2/B, Rac1 and RhoG, WAVE, and WASH is involved but not essential for Salmonella invasion, which is also mediated by a SopB, Rho, RHO kinase, myosin-IIA/B-dependent, actomyosin-mediated contractility (Hanisch et al. 2011). Interestingly, the “zipper” versus “trigger” entry mechanisms should not strictly be attributed to one given species. Indeed, a recent report shows that Salmonella can also enter cells by a zipper-like mechanism mediated by an outer membrane protein named Rck and a signaling pathway that ressembles that of Listeria (Mijouin et al. 2012).
The Invasome of Bartonella: Entry as a Group
Bartonella henselae is a Gram-negative bacterium that specifically colonizes the endothelium (Dehio 2004). It invades and colonizes primary human umbilical vein endothelial cells (HUVEC) by two distinct routes, either as individual bacteria through a “classical” endocytic pathway (which to our knowledge up to now has not been investigated) or as bacterial aggregates that are formed on the cellular surface, followed by their engulfment and internalization via the invasome structure (Dehio et al. 1997; Eicher and Dehio 2012). Attachment to nucleated cells is mediated by nonfimbrial outer membrane adhesion protein belonging to the type V secretion systems (T5SS), for example, trimer autotransporters, including BadA (Bartonella adhesin A) and Vomps (variably expressed outer membrane proteins). Invasome formation and internalization is an actin- and b1-integrindependent process. The massive cytoskeletal rearrangements resulting in invasome-mediated uptake of bacterial aggregates are entirely dependent on the VirB type IV secretion system, but the specific effectors involved are unknown. Interestingly, Bartonella also invades erythrocytes, in this case, with an essential role for the invasion-associated locus proteins (IalA and IalB). This entry probably uses a novel mechanism that deserves investigation.
VIRUSES IN THE ENDOSOME NETWORK
After endocytic uptake, the incoming pathogens enter a complex network of heterogeneous but functionally interconnected endocytic vacuoles and vesicles with early endosomes and macropinosomes as commonly used gateways (Fig. 1). Viruses use the endocytic network of organelles for transit deeper into the cell and for penetration into the cytosol. Unlike some bacteria, they do not modify the composition and functions of the organelles. In early endosomes, the viruses are generally localized in the vacuolar part, sharing this volume with intralumenal vesicles (ILVs). If the particles have not penetrated already in early endosomes, they typically follow the pathway in the direction of late endosomes and endolysosomes (Huotari and Helenius 2011). This pathway involves a complex maturation program that prepares the endosome for fusion with lysosomes (see Klumperman and Raposa 2014; Wandinger-Ness and Zerial 2014). Macropinosomes can also deliver their cargo to lysosomes by fusing with late endosomes, endolysosomes, or lysosomes. Before this can occur, they also undergo a maturation process similar to endosomes. Defects in endosome and macropinosome maturation inhibit the productive entry of many viruses (Khor et al. 2003; Yamauchi et al. 2011; Fuchs and Blaas 2012; Huotari et al. 2012; Krzyzaniak et al. 2013). The majority of viruses are acid-activated, that is, viral membrane fusion proteins and penetration mechanisms depend on exposure of the virus to low pH (Helenius et al. 1980). For viruses that penetrate from early endosomes, the pH threshold is 6 or above, and for late-penetrating viruses it is lower (Lozach et al. 2011a; Vazquez-Calvo et al. 2012a). Receptor interactions and proteolytic processing can also trigger escape with or without acid dependence (Chandran et al. 2005; Simmons et al. 2005; Krzyzaniak et al. 2013). These cues serve as a form of “wake up call” that tell the viruses that they are inside a cell and that it is time for penetration. Much is known about the structure and function of fusion factors that cover the surface of envelope of viruses (Earp et al. 2005). They are oligomeric, type 1 membrane glycoproteins with large ectodomains. The prefusion conformation is metastable, which means that when triggered by low pH or other cues the proteins can undergo major conformational changes and alterations in oligomeric structure. This occurs without the input of additional energy. The changes involve the exposure of hydrophobic or amphipathic peptide sequences (fusion peptides) that allow the protein to insert into the target membrane. After generating a bridge between the two membranes, further conformational changes in clusters of these proteins allows the distance between the membrane to be reduced so that the hydration shell covering the lipid bilayer is disturbed in a focal site. This results in hemifusion (the innermost leaflets of the two bilayers fuse) followed by full fusion, when the outer leaflets also fuse.
BACTERIA IN THE ENDOSOMAL NETWORK
Of intracellular bacterial species, a minority, which includes Listeria, Shigella, Rickettsia, Francisella (Celli and Zahrt 2013), and also Salmonella (Knodler et al. 2010), Mycobacterium marinum (Smith et al. 2008), and Mycobacterium tuberculosis (van der Wel et al. 2007) have evolved like viruses to escape into the cytosol, where they replicate (Table 2). In the case of M. tuberculosis, the intracytosolic localization remains controversial. Intracytosolic bacteria use several molecular tools to escape from the primary internalization vacuoles. The best characterized is the Listeria pore-forming toxin listeriolysin O (Hamon et al. 2012). However, listeriolysin O is not always necessary for escape because listeriolysin O mutants can escape into the cytosol of some human cells, showing that escape can also be controlled by other unknown bacterial and cellular components. In the cytosol, the bacteria subvert cellular defense mechanisms such as antimicrobial peptides, or autophagy. Note that recent data indicate that the induction of autophagy does not necessarily lead to bacterial killing. There are even some bacteria that exploit the autophagic machinery for their own profit (for a recent review, see Mostowy and Cossart 2012). The majority of intracellular bacteria remain in the lumen of endocytic vacuoles, which they modify to provide maximal protection and support (for review, see Kumar and Valdivia 2009). In epithelial cells, these intravacuolar bacteria include Salmonella, Chlamydia, Brucella, Bartonella, and Coxiella. In addition, a few bacteria such as Salmonella, Mycobacteria, Coxiella, and Legionella as well as some noninvasive bacteria are known to survive the killing by macrophages after phagocytosis, and can therefore be found alive inside vacuoles in these phagocytic cells (Fig. 2). The properties of the bacteria-containing vacuoles differ greatly between bacteria. In some cases, the vacuoles acquire or retain properties and markers of early and late endosomal organelles. In others, they have ER-like characteristics and acquire unique properties not shared by organelles of the normal endocytosis pathway. Each bacterial species has evolved sophisticated cell biological mechanisms to ensure that remodeling of the vacuolar environment allows them to survive and replicate. Many of the bacteria devise strategies to ensure that the vacuoles do not mature to a point at which they would fuse with lysosomes. Because each pathogen appears to create a unique niche, we will highlight some of the known characteristics of each vacuole type.
The Salmonella-Containing Vacuole (SCV)
Salmonella replicates in vacuoles with properties of early and late endosomes. The specific characteristics of SCVs are influenced by the SPI-1 T3SS that controls bacterial invasion and the subsequent effects of the SPI-2 T3SS, which is activated 2–3 h after invasion (Yu et al. 2010; Moest and Meresse 2013). The SPI-2 T3SS is responsible for the translocation of approximately 30 effector proteins across the vacuolar membrane, following acidification and nutritional deprivation of the vacuole lumen. The effectors are used for maintenance of the vacuolar membrane and localization of the vacuoles within the host cell, as well as interference with immune signaling and lysosome function. Among the effectors translocated during invasion, SopB is a phosphoinositide phosphatase thought to arrest vacuole maturation by producing a high level of PI3P (thereby maintaining Rab5 localization in the vacuole) (Hernandez et al. 2004) and controlling the membrane surface charge of the SCVs (Bakowski et al. 2010). Subsequently, several other SPI-2 T3SS effector proteins participate in vacuole maintenance through interactions with host factors. For example, SifA interacts with the host scaffold protein SKIP, which interacts with the motor protein kinesin, an interaction required for SCV maturation (Diacovich et al. 2009). Effectors SseG and SseF localize the vacuole close to the Golgi apparatus where the SCV interacts with post-Golgi vesicular traffic (Mota et al. 2009). This may provide the bacteria with nutrients and membrane. SifA also interferes with Rab9-dependent retrograde trafficking of mannose-6-phosphate receptors that deliver lysosomal enzymes from the trans-Golgi network to lysosomes (McGourty et al. 2012). This causes rerouting of the enzymes out of the cell and a concomitant loss of lysosome functionality. This explains why SCVs are devoid of hydrolytic lysosomal enzymes but retain lysosomal membrane glycoproteins in the vacuolar membrane and interact dynamically with the endolysosomal system.
The Chlamydia-Containing Vacuole (the Inclusion)
Soon after endocytosis of Chlamydia, the vacuoles are transported along microtubules to the microtubule-organizing center (MTOC) close to the Golgi complex. They avoid fusion with endosomes and lysosomes. The inclusions interact with several host cell proteins involved in membrane trafficking, fusion, and organelle identity (Bastidas et al. 2013). Several Rabs are recruited including Rab6, 11, and 14. Rab6 and 11 facilitate sphingomyelin transport to the inclusions by regulating fragmentation of the Golgi complex into ministacks (Heuer et al. 2009; Rejman Lipinski et al. 2009). Phosphoinositides are key determinants of membrane identity and vesicle fusion. Several proteins associated with PI4P metabolism (such as OCRL, PI4KIIa, and Arf1) are recruited to the inclusions. In addition to recruiting Rab GTPases, Chlamydia may regulate fusion with host vesicles by recruiting host SNAREs that serve as key factors of the intracellular fusion machinery; among them are two Golgi-specific SNAREs (Syntaxin 6 and GS215) and three endocytic SNAREs (Vamp3, 7, and 8) (Grieshaber et al. 2002). By establishing a close association with the Golgi complex, Chlamydia inclusions intercept sphingomyelin- and cholesterol-containing exocytic vesicles (Hackstadt et al. 1996). They also interact with multivesicular bodies that can act as a source of sphingolipids and cholesterol, with lipid droplets that may serve as a source of neutral lipids, and with mitochondria and lysosomes that may be a source of essential amino acids derived from host protein degradation. Strikingly, elements of the cytoskeleton play an important role in vacuole stabilization (Kumar and Valdivia 2008).
The Legionella-Containing Vacuole (LCV)
Using a type IV secretion system, Legionella prevents fusion of the vacuole in which it resides with endosomal compartments and also blocks autophagy. The vacuole recruits vesicles derived from the ER to create a specialized compartment in which it replicates (Roy and Tilney 2002; Isberg et al. 2009; Hubber and Roy 2010). Fusion is driven by the interaction between a plasma membrane complex consisting of syntaxin and SNAP23 and the ER-localized vSNARE protein Sec22b. In addition to using host SNAREs, Legionella pneumophila encodes SNARE mimics that directly modulate membrane transport (Delevoye et al. 2008; Paumet et al. 2009). Host GTPases that regulate membrane transport are direct targets of Legionella effectors. In particular, Rab1 is recruited to the vacuole, and its activity is controlled by a network of bacterial effectors including DrrA, a GEF protein, and LidA, which interacts with Rab1 on the vacuolar membrane to tether ERderived vesicles that remodel the LCV (Machner and Isberg 2006; Murata et al. 2006).
The Brucella-Containing Vacuole (BCV)
The Brucella abortus replicative vacuole is derived from the endocytic vacuole and matures into an ER-derived organelle (Starr et al. 2008). Brucella-containing vacuoles (BCVs) rapidly acquire several late endosome markers including Rab7 and its effector Rab-interacting lysosomal protein (RILP) (Celli 2006). They are accessible to fluid-phase markers either delivered to the cells or preloaded into lysosomes, indicating that they interact with late endosomes and lysosomes. Intermediate BCVs are acidic and display proteolytic activity up to 12 h postinfection (Celli 2006). Expression of dominant-negative Rab7 prevents conversion of the vacuoles into ER-derived organelles and inhibits replication of Brucella, indicating that BCV maturation requires interactions with functional late endosomal factors (Celli 2006). In the first hours postinfection, Brucella vacuoles merge with late endosomes, with the acquisition of the Lamp1 marker, and the vacuolar pH drops to 4, which is required to activate the VirB type IV secretion machinery. This, in turn, allows the secretion of effectors that prevent the fusion of BCV with lysosomes (Celli et al. 2003). VirB mutants merge with lysosomes where bacteria are killed (Lapaque et al. 2005). Recently, a Brucella protein RicA was shown to recruit Rab2, showing that several Rabs are contributing to the maturation of BCVs (de Barsy et al. 2011; von Bargen et al. 2012).
The Coxiella Vacuole
C. burnetii is the only bacterial pathogen that has evolved to survive and replicate in a lysosome-derived vacuole containing active proteolytic enzymes (Voth and Heinzen 2007). Upon uptake, Coxiella is found in a tight-fitting vacuole positive for early endosomal and autophagy markers (Rab5 and LC3, respectively) (Gutierrez et al. 2005; Romano et al. 2007). The Coxiella-containing vacuoles then merge with late endosomes recruiting markers such as Rab7 (Romano et al. 2007) and acidify, thereby activating the bacterial T4SS (Newton et al. 2013). Coxiella then secretes effectors that drive the generation of a lysosome-derived fusogenic parasitophorous vacuole that occupies the vast majority of the cell cytosol (Comerci et al. 2001; Voth and Heinzen 2007). Upon depletion of intravacuolar nutrients, bacteria enter a latency phase and persist within infected cells, which are protected from apoptosis by a bacterialdriven mechanism (Voth and Heinzen 2007; Beare et al. 2011).
The Mycobacterium tuberculosis Vacuole
M. tuberculosis is internalized in macrophages and is able to arrest the normal maturation of its phagosome. It resides in a vacuole that retains many of the characteristics of a sorting endosome. It has a pH of 6.4 because it lacks the VATPase (Sturgill-Koszycki et al. 1994). However, upon activation of the macrophage, the blocking is overcome and the bacterium is exposed to a lower pH. The bacterium is able to adapt its transcriptional program to this drop in pH (Soldati and Neyrolles 2012; Russell 2013). As said above, an intracytosolic localization of M. tuberculosis remains controversial.
CONCLUDING REMARKS AND FUTURE DIRECTIONS
The reasons why viruses and invasive bacteria make use of endocytosis to enter host cells are fundamentally different. Bacteria seek out intracellular compartments in which they can grow and replicate under optimal conditions. Many remain vacuolar, and some enter the cytosol. Viruses enter cells (the cytosol and the nucleus) because they have no other way to multiply. They need access to the biosynthetic machinery and other services offered by cells. Successful entry of a virus results in the ultimate destruction of the particle, whereas bacteria stay intact and continue to live and divide. The remodeling and reprogramming of endocytic vacuoles are of central importance for many of the invasive bacteria. For viruses, the vacuoles of the endosomal network are merely transient stations on the way to the cytosol. Rather than modifying them, viruses generally count on the normal maturation and function of these organelles for successful entry. Many of the invasive bacteria inject effector proteins into the cytosol of host cells to initiate endocytosis and other changes. Others use receptors that when interacting with their normal ligands are endocytosed via the CME. Viruses are also capable of triggering their endocytosis and other changes in cells, but they do this indirectly using existing cell-surface receptors and existing signaling pathways. Because viruses depend on the cellular machinery for synthesis, they acquire a certain molecular “imprinting” by the host cells that bacteriado not. The lipidsare, forexample, taken from host cell membranes, and carbohydrate moieties reflect the properties of host glycans. Some viruses carry host cell proteins, ribosomes, nucleosomes, histones, and actin filaments as structural components. Although this imprinting can add functionalityand provide camouflage, it can also be used against them by animal hosts, for example, through the detection of the “foreign” origin of incoming arthropod-borne viruses. A long history of coevolution and cohabitation has allowed invasive bacteria and viruses to optimize every aspect of their interaction with host cells. As we have seen, they have become masters at manipulating endocytosis for their own ends. Endocytosis is not only used for entry and replication but also for intoxication by toxins, protection against immune defenses, and cell-mediated movement and transmission between tissues within host organisms. At a first glance, it may seem that each intracellular pathogen has evolved unique solutions. However, given that virtually all depend on the endocytic machinery of the host cell, the options are probably limited. It will be important to expand our cell biological understanding of endocytic pathways and to view already-existing information in this field from the angle of pathogens. The search for cellular targets and new drugs that prevent endocytic uptake and other steps in pathogen entry needs to be better informed by state-of-the-art cell biological input. A better understanding of the endocytic pathways in host cells is likely to provide new options in the development of new antiviral and antibacterial strategies. Development of new therapies against infectious diseases is of utmost importance in a world vulnerable to epidemics and pandemics caused by established and emerging pathogens. Pathogens can also be valuable tools in basic and applied biology. They have been used to study membrane biology, signal transduction, actin-based motility, and so on. Viruses, viruslike particles, pseudotype particles, and mutants, for example, are often used as model ligands in endocytosis research. Latex beads coated with bacterial proteins can also be used. The advantages include the possibility to develop sensitive quantitative assays with the infection itself, providing an easily detected end point. Moreover, it is possible to follow the fate of single pathogens in live cells using light microscopy at increasingly high resolution. Viruses and bacteria are also used as delivery vehicles and carriers for otherwise impermeable substances and macromolecules such as drugs, genes, and proteins.