Many immunologists think about immune responses to virus infection in terms of production of neutralizing antibodies and induction of cytotoxic activity in T lymphocytes and natural killer (NK) cells.
Although these are clearly essential for immunity to viruses that have coevolved with vertebrates (Hedrick, 2004), there are other layers of antiviral defense that operate independently of lymphocytes or NK cells. All metazoan cells possess intrinsic mechanisms to prevent viral replication and spread. In invertebrates such as Drosophila melanogaster, a prominent antiviral resistance mechanism is RNA interference (RNAi) (Galiana-Arnoux et al., 2006; Wang et al., 2006).
In vertebrates, the RNAi pathway is preserved, but its role in antiviral defense appears to have been superseded by a panoply of other mechanisms (Cullen, 2006). These include intrinsic immunity proteins such as Fv or tripartite motif protein (TRIM) 5α , which target capsids of incoming retroviruses, as well as Mx proteins, which target nucleoproteins of bunya- and orthomyxoviruses, and apolipoprotein B mRNA-editing enzyme catalytic polypeptidelike editing complex (APOBEC) 3G, which deaminates retroviral genomes, causing mutations that impair subsequent virus replication (Bieniasz, 2004; Haller et al., 2007) (Figure 1).

Figure 1. Innate Immunity to Viruses Innate responses to viral infection can be focused on viral proteins or viral nucleic acids. Viral proteins are targeted by cytoplasmic restriction factors such as Fv (mouse), TRIM-5α(human), or Mx proteins. Viral nucleic acids trigger PKR, GCN2, and other kinases that phosphorylate eIF2α, which results in the downregulation of translation. In addition, viral RNA stimulates 2' -5' OAS to activate RNase L, resulting in the unspecific degradation of RNA. Both PKR and 2' -5' OAS can additionally signal for the induction of apoptosis. Finally, viral RNA is targeted by the deaminases ADAR1 and APOBEC3G, leading to mutations in viral genomes. Viral nucleic acids and, perhaps, some viral proteins also activate TLRs, RLRs, and DAI to initiate a signaling cascade that promotes activation of IRFs and transcription of IFN-α and - β genes. IFN- α and - β feed back through IFNAR to increase expression of antiviral proteins, thereby amplifying antiviral resistance.
In addition, vertebrate cells possess antiviral mechanisms that are not constitutively active but can be rapidly mobilized by viral presence (Samuel, 2001). For example, viral RNAs activate the kinases protein kinase R (PKR) and/or general control nonderepressible-2 (GCN2), which phosphorylate the a subunit of the translation initiator factor 2 (eIF2α), leading to the downregulation of mRNA translation (Berlanga et al., 2006; Williams, 2001).
Similarly, viral RNA leads to activation of the 2' -5' oligoadenylate synthetase (2' -5' OAS), which activates RNase L, promoting RNA degradation (Player and Torrence, 1998). Finally, infection triggers the activity of adenosine deaminase acting on RNA (ADAR)-1, which deaminates RNA viral replication intermediates (Samuel, 2001) (Figure 1). The shutdown of protein translation, RNA degradation, and RNA deamination are detrimental to cell viability and can result in induction of apoptosis (Samuel, 2001) (Figure 1).
Therefore, it is critical that such antiviral responses only be activated in infected cells. This inducibility implies that all cells possess sensitive, yet specific mechanisms to sense viral presence. In other words, there must be receptors and signaling pathways used to distinguish viral patterns from self. In that sense, PKR, GCN2, 2' -5' OAS, ADAR-1, and other initiators of antiviral immunity (Figure 1) can all be seen as pattern recognition receptors (PRRs) (Janeway, 1989) for viral RNA.
However, the term PRR is generally reserved for those receptors that signal to regulate the transcriptome, for example, inducing expression of proinflammatory cytokines and chemokines that coordinate innate and adaptive immunity (Janeway, 1989). Multiple cytokines are induced by virus infection, including interleukin-6 (IL-6), IL-12 p40, and tumor necrosis factor (TNF), but the hallmark of antiviral responses is the production of type I interferons (IFNs).
Type I IFNs include multiple subtypes encoded by separate intronless genes: 1 IFN-b and 13-14 IFN-a subtypes, depending on species, as well as the lesser-known IFN- ɛ , IFN- κ , IFN- ω , IFN- δ , IFN- τ , and IFN- ζ (Pestka et al., 2004).
In contrast to type II IFN (IFN- γ ), which is made predominantly by T lymphocytes and NK cells in response to T cell receptor (TCR) or NK cell receptor signals, type I IFNs can be produced by all nucleated cells in response to virus infection.
In addition, all cells can respond to type I IFNs through the type I IFN receptor (IFNAR), which binds all subtypes (Stark et al., 1998). Recently, a distinct class of virus-induced IFNs has been identified. These are known as the type III IFNs and include IFN- λ 1, IFN- λ 2, and IFN- λ 3, which are encoded by separate intron-containing genes (Kotenko et al., 2003). The type I, II, and III IFNs all signal via different receptors but share downstream signaling molecules and regulate many of the same genes (Stark et al., 1998).
Studies of mice and humans lacking the signal transducer and activator of transcription 1 (STAT1), a signaling molecule common to all IFN receptors, have revealed that responsiveness to IFNs is absolutely critical for antiviral resistance (Dupuis et al., 2003; Durbin et al., 1996). This is because a major function of IFNs is to increase the expression of the eIF2 α kinases, 2' -5' OAS, ADAR1, Mx, APOBEC, Fv, and TRIM proteins (Samuel, 2001; Stark et al., 1998) (Figure 1). This, in turn, leads to induction of a heightened antiviral state and underlies the discovery of IFNs as virus interference factors 50 years ago (Isaacs and Lindenmann, 1957).
In addition, IFNs act to stimulate NK cells, to amplify dendritic cell (DC) activation, and to facilitate the induction of adaptive immune responses (Le Bon and Tough, 2002) (Figure 1). Therefore, justifiably, the search for receptors and signaling pathways involved in the innate sensing of viruses has centered on those that culminate in transcription of the IFN- α , - β , and - λ genes.
In recent years, there has been rapid progress in this field, and viral PRRs are now known to include members of the toll-like receptor (TLR) family, which also recognizes patterns found in bacteria, fungi, and protozoa (Takeda et al., 2003), and retinoic acid inducible gene I (RIG-I), melanoma differentiation factor-5 (MDA5), laboratory of genetics and physiology-2 (LGP-2), and DNA-dependent activator of IRFs (DAI), all cytosolic proteins which have evolved specifically to sense viruses (Takaoka et al., 2007; Yoneyama and Fujita, 2007). Here, we review our current state of understanding of these innate receptors for viruses and how they function in discriminating virus from self.
Receptors and Signaling Pathways Involved in Type I IFN Induction
Virus Sensing in the Cytosol
Many viruses carry out their entire infectious cycle in the cytosol, but even viruses such as influenza, which replicate in the nucleus, traverse the cytosol on their way in and out of the cell.
Accordingly, cells possess receptors and signaling pathways to induce IFN-α , -β , and -λ gene expression in response to cytosolic viral presence.
Notably, the genes encoding the cytosolic viral PRRs and the components of the downstream signaling pathway are themselves IFN inducible, like those of other antiviral proteins, leading to a positive-feedback loop that can greatly amplify innate antiviral responses (Figure 1).
It has long been thought that this loop is set in motion by the presence of double-stranded RNA (dsRNA) in cells. dsRNA fulfills the criteria for being a marker of virus infection: Long dsRNA molecules are absent from uninfected cells but can be formed by the complementary annealing of two strands of RNA produced during the replication of RNA viruses and as a result of the convergent transcription of tightly packed DNA virus genomes (Jacobs and Langland, 1996).
The first cytoplasmic molecule reported to couple the sensing of dsRNA to IFN- α and - β synthesis was the serine and threonine kinase PKR (Williams, 2001). In some studies, cells lacking functional PKR produced lower amounts of IFN- α and - β when treated with polyriboinosinic:polyribocytidylic acid (poly I:C), a synthetic dsRNA (Der and Lau, 1995; Diebold et al., 2003; Yang et al., 1995). However, this was not the case in response to infection with Newcastle disease virus (NDV) (Honda et al., 2003; Smith et al., 2001) or influenza virus (unpublished data).
Furthermore, although PKR can promote the activation of nuclear factor kappa B (NF-kB) (Kumar et al., 1994), its ability to activate interferon regulatory factors (IRF) 3 and 7, the critical transcription factors in induction of IFN- α and - β via the cytosolic pathway (see below), has yet to be convincingly demonstrated. Therefore, the role of PKR in the induction of type I IFN remains controversial.
RIG-I was recently identified as an IFN-inducible DExD/ H box RNA-helicase that can signal for IRF3 and IRF7 activation and for the induction of IFN -α , -β , and -λ gene expression (Onoguchi et al., 2007; Yoneyama et al., 2004). RIG-I is a cytosolic protein containing an RNA-binding helicase domain and two caspase activation and recruitment domains (CARDs).
The helicase domain has an ATP-binding site, which, when mutated, results in a lossof-function phenotype and dominant negative activity (Yoneyama et al., 2004). RIG-I lacking the CARD domains or bearing mutated CARDs also acts as a dominant negative protein (Yoneyama et al., 2004). Notably, the CARDs of RIG-I are ubiquitinated by the TRIM25 E3 ligase, a modification that is critical for downstream signaling and type I IFN induction (Gack et al., 2007). In contrast, ubiquitination by another E3 ligase, RNF125, promotes the proteasomal degradation of RIG-I and termination of signaling (Arimoto et al., 2007).
Recently, it has been shown that a C-terminal domain within RIG-I acts as an internal repressor of activation (Saito et al., 2007), leading to the notion that RIG-I agonists induce protein multimerization and cause a conformational change that relieves autorepression and exposes the CARDs (Figure 2). Two other poly I:C-binding proteins belong to the RIG-Ilike receptor (RLR) family (Creagh and O’Neill, 2006): MDA5 and LGP-2 (Rothenfusser et al., 2005; Yoneyama et al., 2005).
Like RIG-I, MDA5 bears an RNA-helicase domain and two CARDs, but it lacks the C-terminal repression domain and, consequently, induces the production of IFN- α and - β upon overexpression (Saito et al., 2007).
However, in uninfected cells, the ability of MDA5 to couple to the IRF3 pathway is under negative regulation by dihydroxyacetone kinase (Diao et al., 2007). In contrast, LGP-2 lacks CARDs and blocks IFN- α and - β induction, much like the RIG-I helicase domain, which it resembles (Rothenfusser et al., 2005; Yoneyama et al., 2005). Therefore, LGP-2 has been thought to act as a negative regulator of the two other helicases, a notion that might need revisiting in light of recent analysis of LGP-2-deficient cells (see below).
The discovery of RLRs was rapidly followed by the identification of the downstream adaptor, interferon-b promoter stimulator-1 (IPS-1), also called mitochondrial antiviral signaling protein (MAVS), CARD adaptor inducing IFN-b (CARDIF), or virus-induced signaling adaptor (VISA) (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). IPS-1 contains an N-terminal CARD that forms homotypic interactions with the CARDs of RIG-I and MDA5. This results in activation of the C-terminal catalytic domain and the initiation of a signaling cascade that culminates in the transcription of cytokine genes.
In particular, IPS-1, together with TNF receptor-associated factor (TRAF) 3 and NAK-associated protein 1 (NAP1), promotes the activation of members of the IkB kinase (IKK) family, namely TANK-binding kinase 1 (TBK-1) and IKK3 (Hacker et al., 2006; Kawai et al., 2005; Kumar et al., 2006; Meylan et al., 2005; Oganesyan et al., 2006; Sasai et al., 2006; Seth et al., 2005; Sun et al., 2006; Xu et al., 2005).
The latter phosphorylate and activate IRF7 and/or IRF3 (Fitzgerald et al., 2003; Sharma et al., 2003), which induce the transcription of the IFN- α and IFN- β genes (Honda et al., 2005b). IPS-1 also associates with the adaptor Fasassociated death domain (FADD) and the kinases receptorinteracting protein 1 (RIP1), transforming growth factor b-activated kinase 1 (TAK1), IKKα , and IKKβ to mediate the activation of NF-kB and mitogen-activated protein kinase (MAPK) pathways (Balachandran et al., 2004; Takahashi et al., 2006).
This is important for transcription of the gene encoding IFN- β , whose promoter, unlike the promoters of the Ifna genes, contains NF-kB and activating protein 1 (AP-1) sites in addition to the IRF sites (Honda et al., 2006). Interestingly, IPS-1 is anchored to mitochondria through its C terminus, and the disruption of this association abrogates signaling (Seth et al., 2005) (Figure 2).
The association with mitochondria and signaling through FADD, both of which are involved in apoptosis, suggests a possible crosstalk between IFN-α and -β induction and cell death (Balachandran et al., 2004; Takahashi et al., 2006). Given that both RIG-I and MDA5 bind poly I:C and signal via a common pathway, it was unclear whether they played redundant roles in recognition of viruses. The generation of MDA5- and RIG-I-deficient mice demonstrated a remarkable specificity for the two helicases (Gitlin et al., 2006; Kato et al., 2006).
RIG-I-deficient cells were found to mount greatly diminished IFN- α and -β responses to influenza A virus, vesicular stomatitis virus (VSV), Japanese encephalitis virus (JEV), and Sendai virus (SeV) (Kato et al., 2006), whereas MDA5-deficient cells were selectively unresponsive to picornaviruses, such as encephalomyocarditis virus (EMCV), Theiler’s encephalomyelitis virus, and mengovirus (Gitlin et al., 2006; Kato et al., 2006). Interestingly, the two helicases also differentially affected responses to synthetic dsRNA: IFN-a and -b induction in response to poly I:C was abrogated in cells lacking MDA5 (Gitlin et al., 2006; Kato et al., 2006), whereas RIG-Ideficient cells did not respond to a set of dsRNAs made by annealing complementary ssRNAs generated by in vitro transcription (Kato et al., 2006). Thus, RIG-I and MDA5 appear to recognize patterns from different RNA viruses.
This distinction might not be absolute because responses to JEV and SeV are decreased to a variable extent by MDA5 deficiency (Diao et al., 2007; Kato et al., 2006), suggesting that some viruses might be recognized by both MDA5 and RIG-I. RIG-I can bind poly I:C and polyriboadenylic:polyribouridylic acid (poly A:U), another synthetic dsRNA, (Rothenfusser et al., 2005; Yoneyama et al., 2005) but is not activated by either (Gitlin et al., 2006; Kato et al., 2006), indicating that RNA binding to RLRs is not sufficient for agonist activity. The critical determinant in RIG-I stimulation by RNA was recently identified as the presence of phosphates at the 5' end independently of single or double strandedness (Hornung et al., 2006; Pichlmair et al., 2006).
This has provided a partial explanation for the virus specificity of RIG-I. Thus, the RNA genomes of influenza, rabies virus, and VSV are single stranded but possess 5' triphosphates and therefore can bind RIG-I and promote its activation (Hornung et al., 2006; Pichlmair et al., 2006). Similarly, in vitro transcribed RNA used to make dsRNA (Kato et al., 2006) bears a triphosphate on the first ribonucleotide (Hornung et al., 2006; Kim et al., 2004). In contrast, commercial poly I:C does not generally contain free triphosphates and does not normally activate RIG-I. Notably, the 5' phosphate requirement also explains why RIG-I does not mediate responses to picornaviruses whose genomes are covalently linked at the 5' end to a viral protein that acts as a primer for replication (Racaniello, 2001). The ability of RIG-I to recognize single-stranded RNA (ssRNA) of viral origin challenges the dogma that antiviral responses all rely on the recognition of dsRNA and is consistent with the observation that the replication of negative ssRNA viruses leads to potent IFN responses yet is not accompanied by the formation of immunodetectable dsRNA (Pichlmair et al., 2006; Weber et al., 2006).
Ironically, some of the first investigations into the inducibility of IFN- α and -β had employed ssRNA (Isaacs et al., 1963), although this was later abandoned in favor of the double-stranded version. This is not to say that double strandedness has no role: Intrastrand hybridization clearly contributes to RIG-I recognition, as noted for secondary structures present on the hepatitis C virus (HCV) genome (Saito et al., 2007). However, the notion that linear molecules of viral RNA can contain all the necessary attributes to activate IFN-a and -b responses means that innate detection need not be linked to the generation of complementary RNAs, and explains how viral entry into the cytoplasm can sometimes induce IFN-a and -b responses in the absence of viral replication (Collins et al., 2004; Hidmark et al., 2005; Isaacs and Lindenmann, 1957). It is important to note that we still do not know which viral RNAs are actually sensed by RIG-I during infection. Influenza virus genomic RNA can trigger RIG-I activation in an experimental setting, but it is difficult to envisage how this might happen during infection when the genome segments are coated by viral nucleoprotein and have the viral polymerase bound to their 5' ends (Lamb and Krug, 2001). Furthermore, influenza virus uncoating and replication takes place in the nucleus, a location not subject to surveillance by RIG-I. Therefore, one might expect that RIG-I senses in the influenza virus cycle an RNA intermediate that bears a 5' phosphate and becomes cytoplasmic during infection. Consistent with this notion, during measles infection, RIG-I is activated by gene products that are transcribed early in the virus life cycle (Plumet et al., 2007). In contrast to RIG-I, the agonist for MDA5 remains uncharacterized. Picornaviruses produce abundant dsRNA during cell infection (Pichlmair et al., 2006; Weber et al., 2006), and MDA5 could therefore act simply as a dsRNA sensor, as per the dsRNA dogma. Consistent with this notion, MDA5 binds to and is activated by poly I:C (Gitlin et al., 2006; Kato et al., 2006; Rothenfusser et al., 2005; Yoneyama et al., 2005). However, MDA5 does not compensate for RIG-I deficiency in the response to in vitrotranscribed dsRNA (Kato et al., 2006), indicating that its activation requires more than RNA double strandedness. Notably, among the dsRNA homopolymers, only commercial poly I:C of undefined length induces a potent IFN α and -β response (unpublished data). Therefore, poly I:C, rather than generic dsRNA, mimics the MDA5 agonist generated during picornavirus infection. The role of the third member of the RLR family remains elusive at present. Consistent with an inhibitory role, cells from mice lacking LGP-2 show enhanced production of IFN- α and -β in response to poly I:C treatment or VSV infection (Venkataraman et al., 2007). However, IFN- β production after EMCV infection is actually impaired by LGP-2 deficiency (Venkataraman et al., 2007). Therefore, LGP-2 might play both a positive and negative role in responses initiated by the other two helicases. RNA has been the focus of much attention, but it is not the only nucleic acid that can elicit IFN-a and -b production. Despite thousands of man-years worth of DNA transfections, it was only recently noticed that the introduction of the B helical form of dsDNA into the cytoplasm leads to IFN- α and -β induction (Ishii et al., 2006; Okabe et al., 2005; Stetson and Medzhitov, 2006; Yasuda et al., 2005). The cytosolic dsDNA-sensing pathway converges with the RLR pathway at the point of TBK-1 and IKK3 activation (Ishii et al., 2006; Stetson and Medzhitov, 2006) but does not require the activity of IPS-1, at least in the mouse (Kumar et al., 2006; Sun et al., 2006). One candidate cytosolic dsDNA sensor has recently emerged as the IFN-inducible protein DLM-1 or Z-DNA binding protein 1, now renamed DAI for DNA-dependent activator of IRFs (Takaoka et al., 2007). The binding of DAI to dsDNA increases its ability to interact with TBK-1 and IRF3, and the overexpression of DAI in cells potentiates the response to DNA transfection, whereas the knockdown of endogenous DAI decreases it (Takaoka et al., 2007). It is widely expected that the DNA-sensing pathway will be involved in the detection of DNA viruses and bacteria that access the cytoplasmic compartment.
Virus Sensing in Endosomes
Most viruses encounter the endocytic pathway on their way in and out of cells, either because they infect cells via endosomes or because they bud into those compartments after the completion of their replication cycle (Brandenburg and Zhuang, 2007).
Accordingly, a subset of TLRs appears to be dedicated to surveying endosomes for viral presence. The endosomal TLRs all share the property of being activated by nucleic acids and include TLR3, TLR7, TLR8, and TLR9 (Figure 2). Their expression can be increased by IFN- α and - β , much like that of RLRs and DAI, but endosomal TLRs have a more restricted cellular distribution.
TLR7 and 9 are highly expressed by DCs and, in humans but not mice, by principally those of the plasmacytoid subset (pDC), but can also be expressed by other hematopoietic cells, such as B cells (Iwasaki and Medzhitov, 2004; Reis e Sousa, 2004). TLR3 is expressed more widely, including on nonhematopoietic cells, but it shows preferential expression in nonplasmacytoid (conventional) DCs (cDCs) (Iwasaki and Medzhitov, 2004; Reis e Sousa, 2004).
Less is known about TLR8, which, in the mouse, may be nonfunctional or have a nonimmune role (Jurk et al., 2002; Ma et al., 2006). Notably, although TLR3, 7, and 9 recognize viral agonists in endosomes, the bulk of the receptors at any given time is found in the endoplasmic reticulum in association with the 12 transmembrane spanning protein Unc93b (Latz et al., 2004) (Brinkmann et al., 2007; Tabeta et al., 2006).
The latter plays an essential role in responses mediated by all the endosomal TLRs, possibly by regulating their recruitment to endosomes (Brinkmann et al., 2007; Casrouge et al., 2006; Tabeta et al., 2006).
All TLRs can signal for the activation of NF-kB and MAPK cascades and promote the transcription of multiple cytokine and chemokine genes, but endosomal TLRs have additional pathways to activate IRF1, 3, or 7 and induce IFN- α and -β expression.
TLR3 signals via the adaptor TRIF, which can activate the TBK-1 and IKK ε kinases together with TRAF3 and NAP1, much like IPS-1 (Uematsu and Akira, 2007). This allows TLR3 to couple to the IRF3 pathway in a manner similar to the RLRs and DAI (Figure 2).
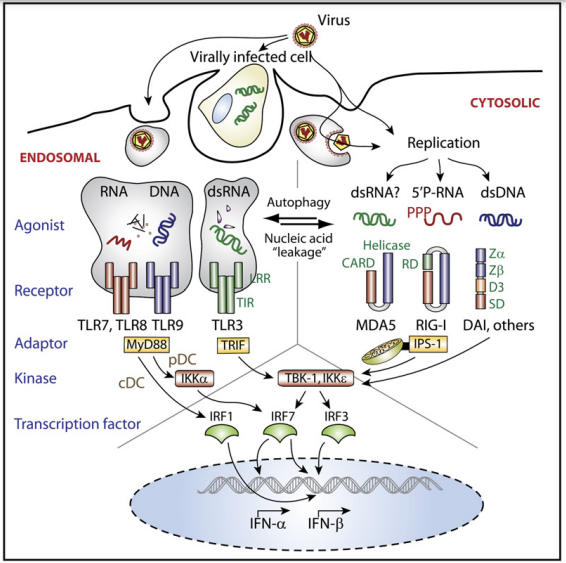
Figure 2. Pathways Coupling Virus Recognition to IFN-α and -β Gene Expression There are two topologically distinct compartments within cells in which viruses can be recognized by PRRs that then signal to induce IFN-α and -b gene transcription. Endosomal pathway: The uptake of viruses or virusinfected cells delivers viral nucleic acids into endosomes, where they are detected by TLRs. In pDC, TLR7, 8, and 9 signal via the adaptor MyD88 and the kinase IKKα to phosphorylate and activate the transcription factor IRF7, which regulates expression of the IFNα and IFN-β genes; in cDC, TLR9 and MyD88 couple to IRF1 but not IRF7, resulting in the expression of IFN-β. TLR3 signals via TRIF, which couples to the kinases TBK-1 and IKKε and phosphorylates IRF3 to induce IFN-α and/or -β. Cytosolic pathway: Viral RNAs are recognized by MDA5 and RIG-I, which signal via the mitochondrion-associated adaptor IPS-1. IPS-1 activates the kinases TBK-1 and IKKε to phosphorylate IRF3 and IRF7 and induce IFN-α and -β gene transcription. Cytosolic DNA receptor(s) such as DAI use an alternative adaptor to couple to TBK-1 and IKKε , and IRF3 and IRF7. Note that the endosomal and cytosolic pathways also couple to NF-kB and MAPK activation, which is important for the expression of the IFN-β gene and regulates the expression of numerous other proinflammatory cytokines and chemokines (not depicted). Note also that there is likely to be extensive crosstalk between the endosomal and cytosolic pathways (depicted by arrows): Nucleic acids can ‘‘leak’’ from endosomes to activate cytosolic PRRs, whereas autophagy can provide cytosolic nucleic acids for endosomal recognition. The following abbreviations and labels are used: leucine-rich repeat (LRR), Toll/IL-1Receptor signaling domain (TIR), caspase activating recruitment domain (CARD), RNA-binding domain (Helicase); RD: repressor domain (RD), Z-DNA binding domains (Zα, Zβ), tentatively named region required for DNA binding (D3), and signaling domain (SD).
In contrast, TLR7 and 9 use a different adaptor, MyD88. The pathways coupling MyD88 to type I IFN gene induction have been studied mostly in the context of TLR9 signaling. TLR9 can be activated by ssDNA oligonucleotides containing unmethylated CpG motifs (Krieg, 2002; Latz et al., 2007). Such oligonucleotides (CpG DNA) induce type I IFN production by pDC through a pathway involving MyD88, IRAK1, and TRAF6 (Uematsu and Akira, 2007). This leads to the activation of IKKa, which, in this setting, substitutes for TBK-1 or IKK3 in phosphorylating and activating IRF7 (Hoshino et al., 2006) (Figure 2).
In cell types other than pDCs, this pathway is not operative, and TLR9 (or TLR7) stimulation only couples to the activation of NF-kB and MAPK, leading to production of cytokines such as IL-6 or IL-12 but not IFN-a. The fact that TLR9 (and TLR7) signaling induces IFN-a only in pDCs has led to the belief that these cells uniquely produce high amounts of type I IFNs when, in fact, cDCs and other cell types also produce large amounts of IFN-a and IFNb through the activation of the cytosolic virus-sensing pathway (Diebold et al., 2003). In addition, mouse cDCs produce IFN-b but not IFN-a in response to TLR9 stimulation through a pathway dependent on IRF1 rather than IRF3 or IRF7 (Figure 2) (Negishi et al., 2006; Schmitz et al., 2007).
Therefore, the defining feature of pDCs is the ability to couple TLR9 and TLR7 signaling to IFN-a gene expression, rather than an ontogenetically determined ability to produce high levels of type I IFNs. Why pDCs couple TLR9 activation to IRF7 whereas cDCs couple the same receptor to IRF1 remains poorly understood, but it might reflect the differential use of cofactors such as osteopontin (Shinohara et al., 2006) and/or the markedly different endosomal routing of CpG in the two cell types. Indeed, the ability of pDC to produce IFN-a in response to TLR9 signaling correlates with their ability to retain CpG DNA in an early endosomal compartment (Guiducci et al., 2006; Honda et al., 2005a). Perhaps because the specialized endosomal traffic in pDCs has evolved to maximize TLR-mediated responses to incoming viruses, these cells have been useful in defining the role of TLR9 and TLR7 in virus recognition. Thus, TLR9 allows pDCs to respond to DNA viruses such as adenovirus, herpes simplex virus (HSV)-1 and -2 or murine cytomegalovirus (MCMV) (Hochrein et al., 2004; Iacobelli-Martinez and Nemerow, 2007; Krug et al., 2004a; Krug et al., 2004b; Lund et al., 2003). Similarly, TLR7 mediates pDC responses to ssRNA viruses such as influenza, VSV, and Sendai virus (Diebold et al., 2004; Lund et al., 2004) and to genomic ssRNA purified from influenza virions or synthetic ssRNA oligonucleotides containing U or GU repeats (Diebold et al., 2004; Diebold et al., 2006; Heil et al., 2004). The latter can also act as agonists for human TLR8 (Heil et al., 2004). Together, these observations have led to a model in which viruses are taken up by pDCs (or other cells) and are subjected to proteolytic degradation in the endosomal compartment, exposing their RNA or DNA genomes for recognition by TLR7, 8, or 9 (Crozat and Beutler, 2004). The process is independent of infection, thereby allowing responses to viruses that do not normally replicate in DCs, as well as to defective viral particles or inactive viruses. The latter could be particularly relevant during infection because viruses neutralized by antibody or complement can be taken up via Fc or complement receptors, thereby potentiating TLR stimulation within endosomes (Wang et al., 2007). Such a mechanism could be especially important to allow the endosomal recognition of viruses that fuse at the plasma membrane. It should be said that there is at present no in vivo evidence that virus recognition by TLR7 and TLR9 operates as per this model. Therefore, it is possible that the actual role of TLR recognition is not in detecting extracellular virus sampled from the external milieu but, perhaps, progeny virions as they bud from the cell or even captured fragments of lysed cells bearing viral particles. An unexpected role for endosomal TLR recognition in viral infection has been found recently, whereby autophagy in pDC allows capture of VSV RNA replication intermediates from the cytosol and recognition via TLR7 (Lee et al., 2007). Autophagosomes fuse with endosomes, effectively bridging the endosomal compartment to the cytosol. Therefore, through autophagy, endosomal TLRs are no longer restricted to sampling the extracellular compartment and can contribute to cytosolic recognition of viruses (Figure 2). It has been argued that pDCs rely on TLR-mediated recognition of viruses, whereas other cells such as cDCs use the cytosolic pathway (Kato et al., 2005). This is not entirely correct; cDCs can use TLR7 and/or 9 to recognize viruses even if this does not result in IFN-a production (see above). Conversely, it is possible that pDCs can use the cytosolic pathway, but this has not been observed in all studies because pDCs are difficult to infect. Notably, Myd88/ pDCs defective in TLR7 and 9 signaling behave like cDCs in that they do not produce IFN-a in response to infection with wild-type influenza virus but do so upon infection with a mutant lacking the NS1 viral protein, which, if present, blocks RIG-I activation (see below) (Diebold et al., 2004). Similarly, Tlr9/ pDCs from murine bone marrow (although not pDCs from spleen) produce IFN-a and -b in response to HSV-1 (Hochrein et al., 2004), and human pDCs produce IFN-a in response to respiratory syncytial virus (RSV) via a cytosolic rather than an endosomal pathway (Hornung et al., 2004). On the other hand, TLR7-deficient murine pDCs do not respond to VSV, perhaps because a high rate of autophagy in those cells sequesters viral RNAs from RIG-I and MDA5 (Lee et al., 2007). TLR3 is the final member of the endosomal TLR family, although it can also be expressed at the plasma membrane in some cell types (Matsumoto et al., 2002). It was originally identified as a receptor for dsRNA on the basis of its ability to mediate responses to poly I:C and to purified reoviral dsRNA (Alexopoulou et al., 2001; Matsumoto et al., 2002). More recently, it has been reported that TLR3 also responds to poly I (Marshall-Clarke et al., 2007), raising the question of whether (as for MDA5) it is truly and simply a receptor for dsRNA. CD14 has been shown to facilitate poly I:C uptake and TLR3 activation (Lee et al., 2006), but the role of TLR3 might not be in recognizing exogenous naked dsRNA, which is unlikely to be found as a free-floating species in the extracellular milieu. Rather, TLR3 might act like TLR7 and 9 in recognizing incoming viruses, such as reoviruses, which have a dsRNA genome, or, through autophagy, it might recognize dsRNA produced by viruses replicating in the cytosol. An additional scenario in which TLR3 has been implicated is in the recognition by DCs of phagocytosed virus-infected cells containing dsRNA (Schulz et al., 2005). In summary, there is a striking parallel between the cytoplasmic and endosomal pathways for the innate sensing of viruses (Figure 2). They both recognize DNA, ssRNA, and dsRNA and signal via parallel pathways for induction of IFN-a and -b. This suggests that the innate immune system focuses on viral nucleic acids as an invariant determinant of viral presence, much like the way in which we use the same determinants for virus classification.
Virus Sensing at the Cell Surface?
In addition to the endosomal TLRs, TLR2 and TLR4 have also been suggested to play a role in sensing of viruses. These TLRs are expressed at the cell surface but can be recruited to the endocytic compartment by internalized ligands (Underhill et al., 1999). TLR2 signaling does not couple to IFN-a and -b induction, although TLR4 does so via the TRIF pathway (Uematsu and Akira, 2007). TLR2 has been shown to respond to components of measles, HCV, MCMV, and HSV (Bieback et al., 2002) (Compton et al., 2003; Duesberg et al., 2002; Kurt-Jones et al., 2004), whereas TLR4 can respond to RSV, retroviruses, and coxsackie B virus (Kurt-Jones et al., 2000; Rassa et al., 2002; Richer et al., 2006). However, when examined closely, the TLR2 and 4 dependence of virus recognition is often restricted to a few virus isolates (Sato et al., 2006). In addition, many examples of TLR2 and TLR4 ‘‘recognition’’ involve viral surface proteins. These often mutate in order to escape adaptive immune recognition by antibodies (Hangartner et al., 2006), so it is hard to envisage why they would not do so to escape TLR recognition. Interestingly, the ability of measles virus to activate TLR2 maps to a single amino acid on the viral hemagglutinin, and this amino acid is preserved in wild-type but not in vaccine strains, suggesting that the wild-type virus actively targets TLR2 (Bieback et al., 2002). Similarly, the analysis of mouse mammary tumor virus infection in TLR4-sufficient and -deficient mice indicates that the virus targets TLR4 to replicate in B cells and to suppress CTL responses (Jude et al., 2003; Rassa et al., 2002). Therefore, some examples of TLR engagement by viruses represent viral exploitation of the TLR system rather than the innate sensing of viruses (Rassa and Ross, 2003).