Introduction
In the past decade, trogocytosis (named from the ancient Greek trogo-, which means gnaw or nibble) has been attracting increasing attention in the vast fields of biology research, including immunology, microbiology, neurology, and developmental biology [1–7]. Trogocytosis is an active process, in which one cell rapidly acquires the fraction of another cell in a cell contact-dependent manner [8] (Figure 1). Trogocytosis is typically distinguished from phagocytosis, which indicates engulfment of the entire cell body (Figure 1).

Figure 1. Characteristics of phagocytosis vs. trogocytosis. (A) In the process of phagocytosis, phagocytes swallow the whole cell body of target cells; (B) in the process of trogocytosis, recipient cells nibble the cell body of donor cells. Trogocytosis results in either (1) the death of target cells (trogocytosis-mediated cell death) or (2) the transfer of cell surface molecules, together with membrane patches, from donor cells to recipient cells (trogocytosis-mediated material transfer).
Trogocytosis was first documented in the field of microbiology in 1979. Brown discovered that Naegleria fowleri, known as “brain-eating amoeba,” nibbles mouse-embryo cells in a piecemeal fashion, a phenomenon termed trogocytosis [9]. In amoeba species, trogocytosis is a means of killing host cells to invade host tissues [10–12]. In the field of immunology, the transfer of major histocompatibility complex (MHC) proteins or immunoglobulins from B cells to T cells was reported in the 1970s [13,14]. In the early 1980s, host-derived MHC proteins were demonstrated to be acquired by donor-derived thymocytes in the system of bone marrow-chimeric mice [15,16]. In 2003, Joly and Hudrisier coined the term trogocytosis to explain the phenomenon of membrane protein transfer after immunological synapse formation in T cells, B cells, and natural killer (NK) cells [8]. To date, trogocytosis has been observed in various types of immune cells, including macrophages [17], dendritic cells (DCs) [18], neutrophils [19], basophils [20], and innate lymphoid cells (ILCs) [21]. Trogocytosis has also been reported in non-immune cells, including lymph node stromal cells [22] and mesenchymal stromal cells [23]. Furthermore, trogocytosis by microglia contributes to brain homeostasis by pruning axons to remodel neurons [24–26].
In the process of trogocytosis, recipient cells acquire membrane fractions, including cell surface molecules, from donor cells. Vanherbergen et al. reported that ~3% of cell surface receptors on donor cells are rapidly transferred to recipient cells in trogocytosis by NK cells [27]. Therefore, recipient cells can gain novel functions by acquiring molecules from donor cells, while donor cells may reduce their cellular functions by the deprivation of their cell surface molecules. Moreover, in some cases, trogocytosis even results in the death of donor cells via a process called trogoptosis [28]. Thus, trogocytosis can modulate the function of both donor and recipient cells [1,2,8,29–32]. In this review, we summarize the immune-modulatory effects of trogocytosis. First, we provide an overview of trogocytosis-mediated cell death. Second, we focus on trogocytosis triggered by ligand– receptor interactions, including T cell receptor (TCR)/MHC and Fcγ receptor/antibodybound cell surface molecule interactions. Finally, we discuss the trogocytosis observed in other immune cells, including basophils, DCs, and innate lymphoid cells.
Trogocytosis-Mediated Cell Death
As described above, trogocytosis by amoebae leads to the death of host cells [6]. Amoebic trogocytosis is mainly studied by using Entamoeba histolytica, a causative microbe of amoebiasis in humans. Ralston et al. reported that amoebae rapidly extract cell fragments from living Jurkat cells via trogocytosis, leading to the upregulation of the intracellular calcium concentration and the death of Jurkat cells [10] (Figure 2A).
Figure 2. Trogocytosis-mediated cell death by amoebae and neutrophils. (A) Amoebae nibble human cells via trogocytosis, leading to the death of human cells. The interaction between amoebae and human cells is dependent on Gal/GalNAc lectin expressed on amoebae; (B) neutrophils nibble antibody-opsonized cancer cells via trogocytosis, resulting in cancer cell death (a process called trogoptosis). Trogoptosis by neutrophils is dependent on FcγR and Mac-1 integrin expressed on neutrophils.
Importantly, amoebae perform trogocytosis-mediated cell killing only when target cells are alive, whereas amoebae perform phagocytosis of target cells when cells are dead [10,11]. Although similar processes are involved in both amoebic trogocytosis and phagocytosis, including Gal/GalNac lectin-mediated cell adhesion, requirement of actin, signaling through phsphoinositol-3 kinase (PI3K), and lysosomal acidification [10,12,33], several reports have implied that the molecular pathways in trogocytosis and phagocytosis are different [6,11]. A recent report demonstrated that AGC family kinase 1 (AGCK1), a molecule downstream of PI3K, is specifically involved in trogocytosis but not in phagocytosis [11], even though AGCK2 is involved in both. Furthermore, a cysteine protease inhibitor impairs amoebic trogocytosis but not phagocytosis, suggesting the possible involvement of a certain amoebic cysteine protease in trogocytosis [34]. Trogocytosis in amoebae would have been discussed in detail by other articles in this issue. Mammalian immune cells can perform trogocytosis-mediated killing of pathogens. Mercer et al. reported that human neutrophils kill Trichomonas vaginalis parasites via trogocytosis [35]. When human neutrophils are co-cultured in vitro with T. vaginalis parasites, neutrophils rapidly nibble the cell body of T. vaginalis, leading to the death of parasites. As observed in trogocytosis by amoebae, human neutrophils trogocytose live parasites, but not heat-inactivated dead parasites. Moreover, trogocytosis-mediated cell death is utilized in antibody-dependent cellular cytotoxicity (ADCC) against cancer cells by neutrophils and macrophages [28,36,37] (Figure 2B). Matlung et al., identified that mouse and human neutrophils nibble antibodyopsonized cancer cells, leading to the necrotic cell death of cancer cells (a process designated as trogoptosis) [28]. Similar to amoebic trogocytosis, neutrophil trogocytosis and cytotoxicity against cancer cells are dependent on PI3K, a signaling molecule downstream of the Fcγ receptor. Trogoptosis by neutrophils is dependent on CD11b/CD18 (Mac-1) integrin-mediated conjugation between neutrophils and cancer cells [28,37]. Interestingly, “don’t eat-me signal” CD47 on cancer cells inhibits neutrophil trogoptosis via interaction with signal regulatory protein a (SIRPa), which provides mechanistic insight into the CD47- SIRPa checkpoint blockade [28,37]. Velumurugan et al., reported a similar trogoptosis phenomenon, in which mouse and human macrophages kill antibody-opsonized cancer cells [36]. Moreover, a recent report revealed that mouse and human vaginal neutrophils can eliminate sperms via trogocytosis, to prevent excess inflammation triggered by exogeneous sperms [38]. Taken together, the mechanisms of trogocytosis-mediated cell death can be operative in both amoeba parasites and mammalian immune cells, suggesting the evolutional conservation of this phenomenon in eukaryotic cells.
Trogocytosis Triggered by LIGAND–Receptor Interaction-1: TCR-Mediated Trogocytosis
3.1. Overview of Trogocytosis Triggered by TCR–MHC Interaction
In most cases, trogocytosis in immune cells requires ligand–receptor interactions [8]. TCR–MHC interaction is a well-characterized one that triggers trogocytosis. Upon stimulation of TCR on T cells by peptide–MHC complexes on antigen-presenting cells (APCs), an immunological synapse is formed between T cells and APCs. An immunological synapse consists of a central supramolecular activation cluster (cSMAC) enriched with TCR and intracellular signaling molecules, and a peripheral supramolecular activation cluster (pSMAC) enriched with integrins that surrounds cSMAC [39]. The formation of an immunological synapse results in the internalization of TCR and the transfer of peptide– MHC (pMHC) complexes, together with membrane fragments of APCs, onto the surface of T cells [40–42]. As observed in trogocytosis by amoebae, TCR-mediated trogocytosis is dependent on both actin polymerization and the TCR signaling pathway, including Src kinase and PI3K [43,44]. Moreover, the formation of an immunological synapse is considered to play a key role in TCR-mediated trogocytosis. Indeed, trogocytosis is abrogated by the blockade of either co-stimulatory molecules (CD28–CD80/CD86 interactions) or integrins (interactions between lymphocyte function-associated antigen 1 (LFA-1) and intercellular adhesion molecule-1 (ICAM-1)) [45]. Interestingly, both co-stimulatory molecules (e.g., CD80, CD86, and OX-40 ligand) and integrin ligands (e.g., ICAM-1) are also transferred, together with pMHC complexes [45–47]. These results imply that a wide variety of molecules present in the immunological synapse are transferred upon TCR-mediated trogocytosis. However, it is worth noting that only limited sets of cell surface molecules are selectively transferred in trogocytosis [48,49]. In general, CD4+ T cells capture peptide–MHC class II (pMHC-II) complexes from APCs, whereas CD8+ T cells acquire peptide–MHC class I (pMHC-I) complexes from APCs. Several reports indicate that CD4+ T cells can acquire both cognate pMHC-II and bystander pMHC-I complexes, possibly because of the localization of bystander pMHC-I in the proximity of cognate pMHC-II complexes [50–52]. Likewise, CD8+ T cells can acquire both cognate pMHC-I and bystander pMHC-II complexes [53]. Martínez-Martín et al. revealed that small GTPases, TC21 and RhoG, play central roles in TCR internalization and the trogocytosis of MHC-II upon immunological synapse formation between CD4+ T cells and APCs [44]. TC21 and RhoG also contribute to the trogocytosis of MHC-I in CD8+ T cells, indicating the essential roles of these small GTPases in TCR-mediated trogocytosis in both CD4+ and CD8+ T cells. Upon formation of an immunological synapse, TC21 and RhoG are internalized, together with TCR. Importantly, CD4+ T cells deficient of TC21 or RhoG fail to internalize TCR and capture MHC-II from APCs, demonstrating their role in TCR-mediated trogocytosis. An in vitro study using Jurkat cells further revealed that RhoG activation in T cells is dependent on TC21 and PI3K. Since TC21 is reported to activate the PI3K pathway [54], one may assume that the immunological synapse induces TC21 activation that, in turn, activates RhoG down-stream of PI3K, leading to the trogocytosis of pMHC complexes.
3.2. The Impact of TCR-Mediated Trogocytosis in CD4+ T Cell Functions
TCR–pMHC-II interaction results in TCR internalization and a subsequent display of pMHC-II on the surface of CD4+ T cells. Several reports have indicated that the trogocytosis of pMHC-II complexes prolongs the association between TCR and pMHC-II complexes, sustains TCR signaling, and promotes the survival of CD4+ T cells, even after the removal of APCs [55–57]. Furthermore, a recent report demonstrated that pMHC-II-dressed CD4+ T cells preferentially differentiate into GATA3+ Th2 cells that secrete IL-4 and IL-5, 72 h after the removal of APCs [57]. Given that weaker TCR signaling drives polarization toward Th2 cells [58], it can be assumed that weak and sustained TCR signaling triggered by acquired pMHC-II may favor Th2 differentiation. However, another report by Boccasavia et al. demonstrated that pMHC-II-dressed CD4+ T cells preferentially differentiate into regulatory T cells (Tregs) after presenting acquired antigens to other naïve T cells [59]. This apparent discrepancy may stem from the difference in the experimental systems they adopted. Further research is required to determine the fate of pMHC-II-dressed CD4+ T cells under more physiological conditions. Several reports have indicated that CD4+ T cells with acquired pMHC-II complexes can further present antigens to cognate CD4+ T cells, leading to CD4+ T cell–T cell interactions [46,59–63]. When MHC-II-dressed CD4+ T cells present antigens to one another, antigen presentation leads to the negative regulation of CD4+ T cell responses [32] (Figure 3). Indeed, Tsang et al., revealed that mouse CD4+ T cells that acquire pMHC-II complexes from APCs induce either the apoptosis or anergy of CD4+ T cells, possibly through mutual antigen presentation among pMHC-II-dressed CD4+ T cells [60]. Helft et al., reported that pMHC-II-dressed CD4+ T cells present antigens to memory CD4+ T cells, which suppresses the proliferation of memory CD4+ T cells [62]. Thus, antigen presentation by pMHC-IIdressed CD4+ T cells form a negative feedback loop that may limit excess T cell responses. On the contrary, Boccasavia et al., recently demonstrated that pMHC-II-dressed CD4+ T cells present antigens to naïve CD4+ T cells, which results in differentiation into pathogenic Th17 cells from naïve CD4+ T cells [59] (Figure 3).
Figure 3. The impact of trogocytosis on CD4+ T cell responses. Upon formation of an immunological synapse, CD4+ T cells acquire pMHC-II complexes from APCs. pMHC-II-dressed CD4+ T cells can present antigens to memory CD4+ T cells, resulting in T cell anergy, while their antigen presentation to naïve CD4+ T cells may result in differentiation into pathogenic Th17 cells. Moreover, trogocytosis by Tregs can reduce the expression of pMHC-II and co-stimulatory molecules on APCs, leading to the suppression of the CD4+ T cell responses elicited by interaction with APCs.
In a model of experimental autoimmune encephalitis (EAE), mice deficient of RhoG, an essential molecule for TCR-mediated trogocytosis, display ameliorated Th17 inflammation, which results in resistance to EAE development. Therefore, antigen presentation by pMHC-II-dressed CD4+ T cells may favor pro-inflammatory Th17 polarization under certain circumstances.
In addition to the impact on CD4+ T cells, TCR-mediated trogocytosis regulates the function of APCs. A recent report by Akkaya et al., revealed that induced regulatory T cells (iTregs) form intense interactions with DCs, leading to a greater capacity for trogocytosis compared to activated CD4+ T cells [64]. Thus, iTregs reduce the expression of cognate pMHC-II complexes on the surface of DCs, resulting in the diminished antigen-presenting capacity of DCs (Figure 3). Moreover, iTregs downregulate the expression of CD80 and CD86 on DCs by means of stripping of these co-stimulatory molecules during trogocytosis [65]. Thymus-derived regulatory T cells (tTregs) capture CD80 and CD86 from DCs via trans-endocytosis mediated by cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), leading to impaired co-stimulation by DCs [66,67]. In addition, CTLA-4 on Tregs reportedly extract co-stimulatory molecules from DCs via trogocytosis [68], suggesting the possible role of trogocytosis in CTLA-4-mediated suppression. Additionally, tTregs reduce the expression of CD70 on DCs via trogocytosis mediated by the interaction with CD27 on Tregs, which results in the inhibition of Th1 priming by DCs [69]. Tregs also downregulate CD137 ligand on DCs via CD137-mediated trogocytosis, leading to the suppression of T cell activation by DCs [70]. Collectively, trogocytosis in Tregs suppresses DC functions via multiple pathways.
3.3. The Impact of TCR-Mediated Trogocytosis on CD8+ T Cell Functions
CD8+ T cells with pMHC-I complexes acquired from APCs via trogocytosis suppress T cell responses through multiple pathways. Huang et al. reported that pMHC-I-dressed CD8+ T cells are sensitive to the cell lysis by other CD8+ T cells, a phenomenon called CD8+ T cell fratricide [40] (Figure 4).
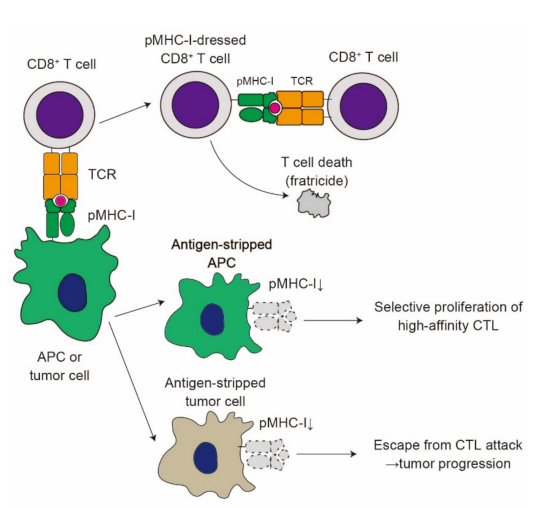
Figure 4. The impact of trogocytosis on CD8+ T cell responses. Upon formation of immunological synapse, CD8+ T cells acquire pMHC-I complexes from APCs or tumor cells. pMHC-I-dressed CD8+ T cells are susceptible to attack from other CD8+ T cells, resulting in cell lysis (fratricide). Trogocytosis reduces the expression of pMHC-I on APCs (antigen stripping). Antigen stripping in APCs favors the selective proliferation of high-affinity CTL. On the contrary, antigen stripping in tumor cells contributes to the escape from CTL attack, leading to tumor progression
A later report demonstrated that CD8+ T cell fratricide can be observed at an extremely high antigen concentration [71], suggesting that the fratricide phenomenon occurs only in the presence of a high antigen concentration. A similar mechanism of fratricide was recently reported in the trogocytosis of T cells bearing chimeric antigen receptors (CARs) [72], which is further discussed below. CD8+ T cells also capture CD80 from APCs through trogocytosis [73]. The acquisition of CD80 by memory CD8+ T cells plays a rather suppressive role in their expansion and IL-2 production, suggesting the regulatory role of trogocytosis in recall immune responses. Trogocytosis by CD8+ T cells takes place either when APCs prime CD8+ T cells or when CD8+ T cells attack target cells, including tumor cells [74,75]. In their interaction with APCs, CD8+ T cells strip pMHC-I complexes from these APCs, which may favor the selective proliferation of CD8+ T cells with high-affinity TCR, leading to affinity maturation of CD8+ T cells [76] (Figure 4). Likewise, CD8+ T cells strip pMHC-I complexes from tumor cells [77]. Chung et al. demonstrated that cytotoxic lymphocytes (CTLs) with high-avidity TCR efficiently lyse melanoma cells, while low-avidity CTLs strip pMHC-I complexes from melanoma cells via trogocytosis, leading to the escape from target cell lysis by high-avidity CTLs (Figure 4). Thus, the trogocytosis of pMHC-I complexes from tumor cells blocks CD8+ T cell responses against tumors. Several reports have demonstrated that CD86 and HLA-G expression on T cells indicates the poor prognosis of multiple myeloma [78–80]. Given that CD86 and HLA-G are transferred from tumor cells via trogocytosis [79], trogocytosis of these molecules by CD8+ T cells may play a suppressive role in tumor immune responses.
Trogocytosis-mediated antigen stripping has recently been reported in CAR T cells [72]. CAR T cells are engineered T cells that express high-affinity variable fragments of antibody (single-chain variable fragment (scFv)) fused with an intracellular domain for signal transduction [81,82]. Therapies using patient-derived CAR T cells display remarkable efficacy against chemotherapy-resistant or refractory B cell malignancies. However, relapse can be observed in a large proportion of patients, and many patients relapse with antigen-low tumors, leading to escape from CAR T cell therapy [81,82]. Hamieh et al., revealed, using a mouse model of leukemia, that the insufficient infusion of CAR T cells causes the trogocytosis of target antigens from tumor cells to CAR T cells [72]. Trogocytosis by CAR T cells reduces the antigen density on target tumor cells, leading to the reduced efficacy of CAR T cell therapy. Moreover, trogocytosis-experienced CAR T cells display an exhausted phenotype and are susceptible to being killed by other T cells (fratricide). These results indicate that tumor cells exploit trogocytosis for escape from CAR T cell therapy.