3. Intracellular Nucleic Acid Sensors
As discussed above, the TLRs play an important role in sensing viral PAMPS that are present within the extracellular compartment, as well as in endosomes. In certain contexts, TLRs can receive viral nucleic acids generated from viruses that replicate in the cytoplasm, via an autophagy mechanism.
A role for intracellular sensors in the clearance of viruses that replicate and reside within the cytosol of cells has recently emerged. Following the generation of mice lacking TLRs and examination of their susceptibility to virus infections, it became clear that additional sensing mechanisms must also exist and contribute to anti-viral defenses. The last decade or more has revealed numerous additional classes of innate sensors.
Of particular relevance to anti-viral defenses was the discovery of specialized classes of cytosolic nucleic acid sensors, termed RIG-I like receptors (RLRs), which recognize intracellular RNA that is introduced to the cytosol during viral infection or that accumulates during replication. Additionally, a diverse selection of intracellular DNA sensors which recognize viral DNA within the cytosol have also emerged.
3.1. The RIG-I like Receptor Family
The RLR family is comprised of three DExD/H box RNA helicases: retinoic acid-inducible gene (RIG-I), melanoma differentiation-associated gene 5 (MDA-5), and laboratory of genetics and physiology-2 (LGP-2) [60–64].
Both RIG-I and MDA-5 are comprised of tandem N-terminal caspase activation and recruitment domains (CARDs) followed by a DExD/H box RNA helicase domain which has ATPase activity and a C-terminal repressor domain (RD). Unlike RIG-I and MDA-5, LGP-2 lacks the N-terminal CARD domains, containing only the RNA helicase domain. As such, LGP-2 was postulated to act as a negative regulator of the other RLRs [61,63].
Under resting conditions, RIG-I resides in the cytoplasm in an inactive form that is auto inhibited by its regulatory domain. Upon viral infection, RIG-I undergoes a conformational change by which it dimerizes in an ATP dependent manner [63]. The activated multimeric form of RIG-I or MDA5 then interacts with the downstream adaptor protein mitochondrial antiviral signaling protein (MAVS), also known as VISA, IPS-1, and CARDIF, via CARD-CARD interactions.
MAVS is localized to the outer leaflet of the mitochondrial membrane, which is an essential location to support downstream signaling. Recently, MAVS was also shown to be localized on peroxisomes, from where it induces an early antiviral response through the direct induction of a subset of anti-viral genes via the transcription factor IRF1.
Upon engagement of RIG-I or MDA5 with MAVS, MAVS activates the IKK-related kinase, TBK1/IKKi, which activates IRF3/IRF7, resulting in the transcription of type I interferons. MAVS also activates NF-κB through recruitment of TRADD, FADD, caspase-8, and caspase-10 [65–69].
3.2. RNA Recognition by RLRs
The RLRs are critical components of the anti-viral defense pathway in many cell types including fibroblasts, epithelial cells, and conventional dendritic cells [70].
Initially, it was thought that both RIG-I and MDA-5 recognized the synthetic dsRNA, polyinosinic acid (polyI:C). However, studies from RIG-I and MDA-5 deficient mice determined that MDA-5 alone was responsible for interferon production by polyI:C stimulation [71].
Instead, RIG-I recognizes 5‟-triphosphorylated, uncapped ssRNA, which is a common feature in many viral genomes. However, it is unable to recognize the capped 5‟-ppp ssRNA from the host cell [72–74].
These finding suggest that RIG-I uses the 5‟ end of a transcript to discriminate between viral and host RNA. MDA-5 distinguishes between viral and host RNA not by its 5‟ end, but rather by the length of the RNA sequence; long dsRNA is not naturally present in host cells and acts as a ligand of MDA-5. In addition to recognizing 5‟-triphosphate RNA, RIG-I is also capable of recognizing short dsRNA, which is produced as a byproduct of viral replication [75].
RIG-I and MDA-5 appear to differentially recognize different classes of RNA viruses. Studies involving RIG-I deficient mice implicated RIG-I in the recognition of vesicular stomatitis virus (VSV), rabies virus, SV, Newcastle disease virus (NDV), RSV, measles virus, Influenza A and B, hepatitis C virus (HCV), Japanese encephalitis virus, and ebola virus [53,70,71,76–78]. Studies from MDA-5 deficient mice show that MDA-5 is able to recognize EMCV, theiler‟s virus, and mengo virus [71,77].
All of these viruses do not contain a 5‟ triphosphate RNA, but are able to produce long dsRNA, providing further evidence that MDA5 discriminates between self and non-self RNA based on sequence length and not the 5‟triphosphate.
More recently studies have shown that both CVB and poliovirus are dependent on MDA-5 for type I IFN production [79,80]. Moreover, some viruses, such as dengue, West Nile virus, and reovirus, signal through a combination of both RIG-I and MDA-5 [79,81,82].
As discussed above, LGP-2 lacks N-terminal CARD domains, and was first thought to be a negative regulator of RLR function [61,63]. Initial studies found that overexpression of LGP-2 decreased the capacity of SV and NDV to induce interferon production.
Evidence that LGP-2 could associate with RIG-I through mutual RD domains led to the proposal that LGP-2 directly prevented RIG-I association and activation. Consistent with this idea, interferon signaling was found to be increased in LGP-2 deficient mice responding to polyI:C, providing evidence for negative regulation of MDA-5 as well [83].
A second in vivo study using LGP-2 deficient mice as well as mice harboring an inactive ATPase in the DExD/H-box RNA helicase domain showed that LGP-2 acted as a positive regulator of RIG-I and MDA-5-mediated signaling after infection by RIG-I and MDA-5-specific RNA viruses. This phenotype is consistent with the possibility that LGP-2 might promote RNA accessibility, thus enabling RIG-I or MDA-5 dependent viral recognition. Further studies on these mice will no doubt clarify this upstream mechanism and the role of LGP-2 in this pathway.
3.3. DDX3
Another member of the DExD/H box RNA helicase family, DDX3, has also recently been implicated in anti-viral defenses. Schroder et al. found that the vaccinia virus protein K7 inhibited IFNβ induction by binding to DDX3, which led to the discovery that DDX3 had a positive role in the RLR signaling pathway [84].
A more recent study reported that DDX3 binds to both polyI:C and viral RNA introduced into the cytosol and associates with MAVS/IPS-1 to upregulate IFNβ production. These results led the authors to speculate that DDX3 might enhance RNA recognition, forming a complex with RIG-I and MAVS to induce interferon production [85]. Further studies are required to determine whether DDX3 is a bona fide RNA sensor or a component of the RLR signaling pathway in order to fully understand the function DDX3 plays in anti-viral surveillance and signaling.
3.4. Cytosolic DNA Sensors
Prior to the discovery of TLR9, it was known that DNA derived from pathogens could activate fibroblasts to produce type I IFNs [86]. This phenomenon was ignored or underestimated for decades and was rediscovered following the observation that transfection of pathogen-derived dsDNA activated a TLR9 negative thyroid cell line to upregulate various immunological genes [87].
Akira and colleagues subsequently demonstrated that TLR9−/− MEFs, which failed to respond to CpG DNA, produced large amounts of IFN in response to transfection with synthetic b-form dsDNA or genomic DNA isolated from bacteria, viruses, and mammalian cells [87]. This was similar to findings presented by the Medzhitov lab using a 45 bp dsDNA region from the Listeria monocytogenes genome.
Cytosolic administration of dsDNA did not appear to utilize any known TLRs to induce interferon since cells from mice lacking both MyD88 and TRIF responded normally. Like the cytosolic RNA recognition pathways, cytosolic DNA recognition also leads ultimately to activation of TBK1 and IRF-3 and production of type I IFNs. However, the signaling pathway linking upstream DNA sensors to TBK1 are poorly characterized. TBK1 associates with DDX3, a DEAD box RNA helicase, which regulates IFNβ transcription via IRF-3 [84,85]. In addition, TBK1 interacts with the exocyst protein Sec5 in a complex that includes the recently identified endoplasmic reticulum (ER) adaptor stimulator of interferon genes (STING) [69,88–90]. STING plays a central role in the signaling pathway upstream of TBK1 following HSV infection [69]. STING also interacts with the ER translocon components Sec61β and TrapB in a manner essential for regulation of cytosolic DNA-induced type I IFN production, although the mechanistic understanding of this finding is not known [88]. In unstimulated cells, STING localizes to the ER and perhaps ER-associated mitochondria [90]. Following stimulation with cytosolic DNA and HSV-1, STING translocates to perinuclear foci, via the Golgi [88]. STING localizes partially to endosomes, particularly Sec5 positive structures [88], whilst another report has demonstrated that STING localizes to vesicular structures, which are not peroxisomes, mitochondria, endosomes or autophagosomes [91]. Further work is required to clarify the precise subcellular localization of STING. What is clear is the essential role of STING in cytosolic DNA sensing pathways. Much less clear is the mechanisms or receptors which act upstream of STING. A growing number of DNA sensors have now been implicated and will be outlined below.
3.5. DAI
DNA-dependent activator of IFN-regulatory factors (DAI) was among the first of the cytosolic DNA sensors to be discovered. It is composed of two binding domains for left-handed, Z form DNA, although the protein can recognize B form DNA as well. When DAI was exogenously expressed in L929 cells, it increased type I IFN production in a dose dependent manner following stimulation by both B and Z form DNA. Similarly, knockdown of DAI with siRNA impaired type I IFN production in response to DNA, the 45 bp interferon stimulatory DNA (ISD) from Listeria and the herpesvirus, HSV-1 [92,93]. The production of type-1 interferons by fibroblasts in response to HCMV was also found to be dependent on DAI [94]. DAI-knockout mice were subsequently generated, and surprisingly, cells derived from DAI deficient mice respond normally to synthetic and viral dsDNA [92,95]. These results suggested that DAI might play a cell type specific, and redundant role in sensing cytoplasmic DNA, and that other sensors must also be necessary for inducing these responses.
3.6. RNA Pol III
As discussed above, both synthetic and viral RNA trigger the production of type I IFNs via RIG-I. Although, the RLRs are sensors of RNA, some data has suggested a role for this system in detection of DNA. A somewhat surprising finding was that synthetic B-form dsDNA can also induce IFN β production in human cells in a manner that was dependent on the RIG-I adapter molecule MAVS [52–54]. These findings suggested the existence of an unknown DNA sensor that would signal via MAVS. Recently, two independent studies have provided an explanation for these findings and shown that AT-rich DNA can be transcribed by RNA polymerase III into 5'-ppp RNA, which subsequently activates RIG-I [52,55]. This pathway was reported to be involved in type I IFN induction during EBV infections where the EBERs are transcribed by RNA polymerase III [56]. This indirect DNA-sensing system was also reported to be involved in induction of type I IFN following HSV-1 or Legionella infection [52,55,57].
3.7. LRRFIP1
In addition to DAI and RNA Pol III, Leucine-rich repeat flightless-interacting protein 1 (LRRFIP1) has recently been implicated as a regulator of DNA-driven innate immune signaling. LRRFIP1 was found to bind to the drosophila homolog flightless I and play a role in actin organization during drosophila embrogenesis. In a study using Listeria monocytogenes to screen for potential cytosolic DNA sensing molecules, siRNA against LRRFIP1 was found to inhibit type I IFN production induced by the bacteria. The authors showed that the IFN response to VSV was dampened in these cells as well. Furthermore, knockdown of LRRFIP1 inhibited IFN production in response to polyI:C, and the synthetic DNA species, poly(dG:dC) and poly(dA:dT), implicating LRRFIP1 in the recognition of both dsRNA and both B and Z form dsDNA. Surprisingly, this function is independent of RNA Pol III. LRRFIP1 does not regulate IRF3 activation but instead appears to regulate a novel β-catenindependent coactivator pathway. LRRFIP1 binds RNA or DNA and leads to phosphorylation of β-Catenin, which subsequently translocates to the nucleus where it associates with the p300 acetyltransferase at the IFNβ1 promoter, leading to increased IFNβ production [101]. Although LRRFIP1 has been implicated in the recognition of both Listeria monocytogenes and VSV, further studies are needed in order to determine its role in sensing other viruses, particularly DNA viruses.
3.8. IFI16
While analyzing immune responses to a dsDNA region derived from the VV and HSV-1 genomes, Bowie et al. identified IFI16 as a DNA binding protein which interacted with these dsDNAs. IFI16 is a member of the PyHIN (pyrin and HIN200 domain-containing) protein family. The PHYIN family consists of 4 family members: IFIX, IFI16, MNDA and AIM2. All contain one or more HIN200 domains, which recognize DNA as well as a pyrin domain. Knockdown of IFI16 or p204 (a member of the murine PYHIN family) led to a reduction in IFNβ responses to these dsDNAs while responses to the RNA virus SV was unaffected. Although IFI16 is primarily nuclear in most cell types, in macrophages IFI16 also localized to the cytosolic compartment where it co-localized with dsDNA introduced via lipofectamine. Association of IFI16 with STING was required for the production of IFNβ in response to these DNA motifs. siRNA knockdown of IFI16, and its mouse homolog p204 led to a decrease in IRF3 and NF-κB activation and IFNβ gene induction following infection of cells with HSV-1 [102].
3.9. DDX9 and 36
Also in the family of DExD/H box RNA helicases, DHX9 and DHX36 have recently been shown to recognize and bind CpG-B and CpG-A DNA, respectively in plasmacytoid dendritic cells. Activation of DHX9 leads to IRF-7 activation and IFNα production, while activation of DHX36 leads to the activation of NF-κB and the production of IL-6 and TNFα. siRNA knockdown of DHX9 and DHX36 inhibited cytokine production in response to the DNA virus HSV-1, while response to the RNA virus influenza A was unaffected [103].
4. Inflammasomes
Although the sensing of cytoplasmic DNA is linked to the transcriptional induction of type I IFN and other pro-inflammatory cytokines, cytosolic DNA has also been shown to trigger the caspase-1-dependent maturation of the pro-inflammatory cytokines IL-1β and IL-18 [104,105].
IL-1β, a close biological relative of TNFα, is involved in innate cell recruitment, activation of T-lymphocytes and induction of fever [106]. IL-18 increases the cytolytic activity and IFNγ production of natural killer (NK) cells and influences neutrophil recruitment and activation [106,107]. Growing evidence supports the importance of these cytokines in anti-viral defenses [108,109].
Mice lacking either one of these cytokines have demonstrated enhanced susceptibility to influenza A virus and HSV-1 infections [110]. Moreover, pretreating mice with IL-18 protects them from subsequent HSV-1 and VV challenge [111,112]. In contrast to type I IFNs and TNFα, the production of IL-1β is controlled at the level of transcription, translation, maturation and secretion [113,114].
Many cell stimuli including TLR-ligands activate the transcription of the pro-forms of IL-1β and IL-18. Unlike most other cytokines however, these pro-cytokines lack leader sequences and are retained in the cytoplasm rather than loaded into secretory vesicles. Maturation (i.e., the cleavage) of pro-IL-1β and pro-IL-18 is catalyzed by the cysteine protease caspase-1 (formerly known as IL-1 converting enzyme).
In resting cells, caspase-1 itself is present as an inactive zymogen pro-caspase-1 [115]. A large 'inflammasome protein complex' controls the activity of the inflammatory caspase-1 [115].
Several protein complexes have been shown to form inflammasomes upon recognizing specific stimuli. NLRPs 2 to 14, which contain a C-terminal LRR-rich domain, a central nucleotide-binding NACHT oligomerization domain, and an N-terminal protein–protein interaction pyrin domain (PYD) associate with the PYD containing adaptor molecule apoptosis-associated speck-like protein (ASC; also termed pycard or TMS1) [116].
ASC links the NLRP's via its C-terminal CARD domain to the CARD domain of pro-caspase-1. This close association of pro-caspase-1 molecules is then believed to provoke self-cleavage into active caspase-1. Active caspase-1 then cleaves pro-IL-1β and pro-IL18. ASC is critical for caspase-1 activation in response to many stimuli [106,107,115,117,118].
4.1. AIM2
Cytosolic dsDNA also triggers an ASC dependent activation of caspase-1 resulting in the maturation and secretion of IL-1β and IL-18. These findings suggested the existence of an inflammsome complex that can be triggered by DNA. Analysis of this response in macrophages lacking members of the NLRs revealed normal caspase-1 activation in these cells. Subsequent studies from several groups revealed that this response was instead dependent on AIM2 (Absent in melanoma-2), an interferon inducible protein that belongs to the same PYHIN family as IFI16 [105,119–121]. AIM2 recognizes cytosolic dsDNA of self and nonself origin including viral DNA via its HIN200 domain in a sequence-independent manner. Contrary to other cytosolic sensors of DNA, the recognition of DNA by AIM2 triggers the assembly of an inflammasome complex. Upon DNA binding, AIM2 likely undergoes oligomerization and associates with ASC via homotypic pyrin-pyrin domain interactions, which in turn recruits pro-caspase 1. Published data has shown that the AIM2 inflammasome is an integral component of innate sensing of DNA viruses [109]. AIM2 is essential for the activation of caspase-1 and proteolytic processing of IL-1β and IL-18 in antigen presenting cells in response to infection with MCMV and VV. Furthermore, AIM2-ASC dependent IL-18 secretion and NK-cell activation is critical in the early control MCMV infection in vivo [105,109]. In addition to viruses, AIM2 has also been shown to recognize Francisella tularensis and as observed for DNA viruses appears to be critical in early control of Francisella tularensis infection in vivo. Moreoever, AIM2 as well as NLRP3 and IPAF function in a redundant manner in the recognition of Listeria monocytogenes [109,122].
4.2. NLRP3
In addition to the AIM2 inflammasome, a number of recent studies have shown that mice deficient in NLRP3 are more susceptible to virus infections, particularly RNA viruses [104,123,124]. Loss of NLRP3 was found to attenuate the normal IL-1β and IL-18 responses to influenza virus and was associated with diminished innate cell recruitment to the lung and increased pathology [123]. Further studies revealed that influenza‟s M2 protein, a proton-specific ion channel was needed to trigger the NLRP3 inflammasome [124]. Viral RNA has also been shown to trigger NLRP3 activation, although this is unlikely to be a direct RNA-NLRP3-interaction. The precise relationship between M2 and RNA in NLRP3 activation remains to be clarified. The NLRP3 inflammasome also plays a role in the response to adenovirus, a DNA virus [104]. Peritoneal macrophages isolated from NLRP3 or ASC deficient mice exposed to adenovirus are unable to secrete mature IL-1β [104]. When challenged in vivo, NLRP3 knockout mice had reduced levels of IL-1β, IL-6, CCL4 (MIP-1β) and CXCL10 (IP-10) in the liver. Recently, a viral NLR homolog was identified in the dsDNA virus, KSHV. The KSHV tegument protein ORF63 appears to be an NLR homolog that can inhibit inflammasome activation by binding to NLRP1 and NLRP3 [58]. Inflammasome activation suppresses KSHV reactivation from latency, suggesting that inflammasome activation and IL-1 β mediated signaling facilitates KSHV latency. These observations are consistent with a model whereby the KSHV tegument ORF63 protein might bind NLRP3 and/or NLRP1 to block the detrimental effects of inflammasome activation. Intriguingly, a recent study has revealed a role for IFI16 in the recognition of Kaposi sarcoma-associated herpesvirus (KSHV) in endothelial cells. IFI16 is known to recognize viral DNA in the cytosol and drive type I Interferon production, as discussed above. In endothelial cells however, IFI16 in the nucleus can sense the KSHV DNA and form a complex with the inflammasome adapter molecule ASC. These findings suggest that IFI16 can form an inflammasome complex following recognition of nuclear DNA during infection with this virus [59]. Figure 2 portrays the cytosolic and nuclear receptors known to respond to viral pathogens and their downstream signal pathways.
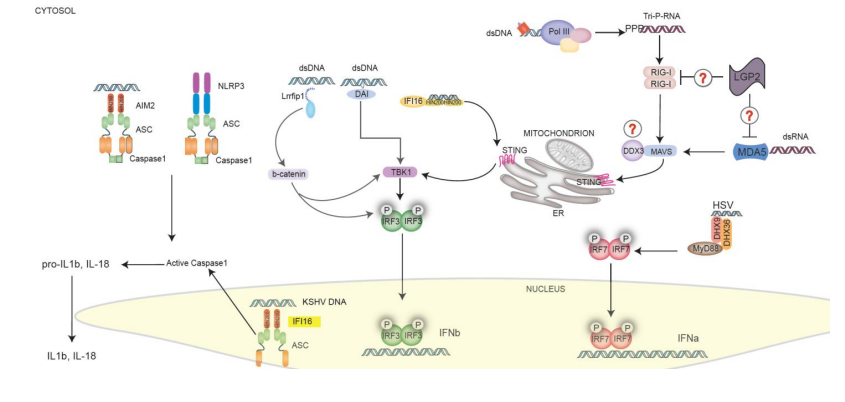
Figure 2. Cytosolic and Nuclear Pattern Recognition Receptors (PRRs). A multitude of DNA sensors, including IFI16, RNA Polymerase III, DAI, LRRFIP1, and DDX9/36 recognize DNA and drive type I IFNs and cytokine production. RIG-I and MDA5 recognize RNA in the cytosol. All of these molecules converge on STING in the case of DNA or MAVS in the case of RNA. STING and MAVS then engage either the TBK1-IRF3 or the IKKb-NFkB pathways, resulting in the activation of type I IFN responses and inflammatory cytokines, respectively. AIM2 (which binds to dsDNA) and NLRP3 (which can respond to viral RNA (probably indirectly)) act in the cytosol to promote the formation of a multiprotein inflammasome complex that contains the adaptor protein ASC, and caspase-1. IFI16 can also detect DNA in the nucleus during KSHV infection. Nuclear IFI16 engages ASC which then triggers caspase-1 in the cytosol. Activation of caspase-1 results in the proteolytic cleavage of pro-IL-1β and pro-IL-18 to IL-1β and IL-18, respectively. The mature cytokines can then be released from the cell.
5. Conclusions and Future Perspectives
Over the past decade our understanding of how the innate immune system detects viruses and triggers antiviral responses has increased immensely. Our knowledge of what constitutes a PAMP, once limited to classical TLR activators such as LPS, has recently expanded to include nucleic acids. This has led to the discovery of a variety of cytosolic RNA and DNA receptors and their downstream signaling pathways. Although our grasp of TLR function has matured significantly over the past decade, a number of prominent questions remain regarding cytosolic and nuclear PRR signaling. First, many of the cytosolic sensors appear to play redundant roles in viral detection. Such overlapping defense strategies may have evolved in order to combat viral evasion mechanisms. Defining the function of newly identified PRRs in immune defense to viral infection is an important step in understanding their unique or ancillary contributions to pathogenesis. Secondly, it remains unclear how some nucleic acid sensors discriminate self from non-self. Just as RIG-I recognizes the 5‟ triphosphate moiety found principally on viral RNAs, a mechanism presumably exists allowing PRRs such as IFI16 to distinguish between virally derived and host DNA. Another question that must be addressed is how viral RNA and DNA is made accessible to PRRs. For instance, it is not well understood how nucleic acids are presented to cytosolic sensors in cases such as HSV infection where viral DNA is shielded by a capsid in the cytoplasm and replicates within the nucleus. As we explore these and other questions it is imperative that we apply our findings in human model systems. By encouraging cooperation between basic and clinical communities we can ensure that new discoveries are quickly translated into therapeutic strategies.