1. Introduction
Cells of the innate immune system utilize pattern recognition receptors (PRRs) to identify viral pathogens by engaging pathogen-associated molecular patterns (PAMPs). Once thought to be moieties found only on pathogens our understanding of PAMPs (pathogen associated molecular patterns) has expanded to include not only classical PAMPS such as lipopolysaccharides found on bacteria but also nucleic acids.
Nucleic acid sensing has emerged as a major component of the immune systems anti-microbial arsenal. A diverse range of pathogens are sensed via recognition of their genomes or nucleic acids which accumulate during their replication. Nowhere is this more prevalent than in viral detection.
PRRs respond to signatures present in viruses such as 5‟ triphosphate RNA, which is not normally found in host RNA or to nucleic acids such as viral DNA which is exposed to sensors localized in the cytoplasm. Of the PRRs, the Toll-like receptors (TLRs) are perhaps the most extensively studied. TLRs are type 1 transmembrane proteins that traffic between the plasma membrane and endosomal vesicles.
They are primarily responsible for detecting PAMPs in the extracellular environment. Those located on the plasma membrane are usually specific for hydrophobic lipids and proteins while those found in endosomes detect nucleic acids. This segregation appears intentional allowing innate cells to respond to components of the viral envelope such as fusion machinery at their surface. In contrast, nucleic acids are detected in the endosome where many viruses uncoat their genomes and enter the cytoplasm.
Upon reaching the cytoplasm, viral components are subject to the scrutiny of the retinoic acid-inducble gene I-like receptors (RLRs), the nucleotide oligomerization domain-like receptors (NLRs) and cytosolic DNA sensors such as members of the AIM2 family.
Similar to TLRs, RLRs and DNA sensors regulate transcription factors essential for the production of interferons and cytokines. In contrast, NLRs and AIM2 are mainly responsible for the maturation of IL-1β and IL-18 through the activation of caspase-1. Interestingly, the immature forms of IL-1β and IL-18 are induced by TLR signaling while NLRs act as a "checkpoint", regulating the activation and release of these potent effectors. In addition to the production of proinflammatory molecules, many classes of PRRs mobilize the adaptive immune response by increasing expression of MHC class II and inducing expression of the costimulatory molecules CD40, CD80 and CD86.
2. The Toll-like Receptors
The Toll protein was first recognized for its role in dorsal-ventral patterning of Drosophila embryos. Later studies found it to be important for the adult fly‟s immune response to bacterial and fungal infections fueling the search for mammalian homologs. To date, 10 TLRs have been identified in humans, 13 in mice with TLRs 1-9 common to both.
TLR1, TLR2, TLR4, TLR5 and TLR6 are located on the plasma membrane while TLR3, TLR7, TLR8, and TLR9 are endosomal.
All TLRs share a common architecture consisting of extracellular leucine-rich repeats and a cytoplasmic Toll/Interleukin-1 Receptor (TIR) domain [1].
These receptors signal as dimers, differentially recruiting the adaptor proteins Mal (MyD88 adapter-like), also called TIRAP (TIR domain-containing adaptor protein) and MyD88 (Myeloid differentiation primary response gene 88) and/or TRIF (TIR-domain-containing adaptor inducing IFNβ) and TRAM (Trif-related adaptor molecule) [1].
Adaptors initiate signal cascades culminating in the activation of nuclear factor kappa b (NF-κB), mitogen-activated protein kinase (MAPK) and interferon regulatory factors 1, 3, 5 and 7 (IRF-3, -5 and -7) [2]. Together these transcription factors not only drive expression of interferons, cytokines and chemokines but also influence cellular maturation and survival.
2.1. TLR Signaling
With the exception of TLR3 all TLRs recruit MyD88 upon activation. In the case of TLR2 and TLR4, the Mal/TIRAP protein acts as a bridging adapter to recruit MyD88 to the activated receptor [3].
MyD88‟s death domain associates with and activates IL-1R-associated kinase 1 (IRAK-1) and/or IRAK-2. IRAK-4 also transiently interacts with this complex and is thought to phosphorylate IRAK-1. IRAK-1 is subsequently released and engages TNFα receptor-associated factor 6 (TRAF6). Activated TRAF6 is capable of K63-linked polyubiquitination of itself and other proteins.

It interacts with NF-κB essential modulator (NEMO, also known as IKKγ), another of its ubiquitination targets, as well as TGF-β-activated kinase-1 (TAK1) and the TAK1 binding proteins (TAB1, TAB2 and TAB3). NEMO forms a complex with IKKα and IKKβ, which are the catalytic kinases responsible for phosphorylating IκB. IκB binds to and sequesters NF-κB in the cytoplasm.
Phosphorylation 인산화
Following phosphorylation, IκB is ubiquitinated and finally degraded by the proteasome releasing NF-κB to enter the nucleus and induce gene expression. Studies indicate that TAK1 plays an essential role in both the NF-κB and MAPK pathways by phosphorylating IKKβ and c-Jun N-terminal kinase (JNK), respectively [4,5].

TLR3 is incapable of recruiting MyD88 and instead interacts with the adaptor protein TIR-domaincontaining adapter-inducing interferon-β (TRIF). TRIF can directly bind TRAF6 and induce NF-κB in a manner similar to MyD88.
In contrast to MyD88, TRIF is also able to recruit the protein receptor interacting protein-1 (RIP-1). RIP-1 synergizes with TRAF6 resulting in more potent NF-κB activation.
A third protein recruited to TRIF is TRAF3. TRAF3 associates with TANK binding kinase-1 (TBK1) and IKKi and is essential for the production of type I interferon. TBK1 and IKKi mediate this production by phosphorylating interferon regulatory factor-3 (IRF3) and IRF7. This allows them to dimerize and enter the nucleus where they cooperate with NF-κB and activator protein 1 (AP-1) to bring about target gene transcription.
TLR4 can recruit TRIF through the adaptor TRIF-related adaptor molecule (TRAM) and can therefore signal through either pathway. A number of primary immunodeficiencies in humans are the result of defects in the innate signal pathways described above. For instance, one study of children with nonfunctional MyD88 proteins found they were predisposed to recurrent life-threatening pyogenic bacterial infections [6].
A similar phenotype has been reported in patients with IRAK-4 deficiency [7]. A study of two unrelated children with defects in UNC-93B1, a protein thought to be involved in trafficking TLR3, TLR7, TLR8 and TLR9 to the endosome, found an increased susceptibility to encephalitic herpes simplex virus-1 infection [8]. PBMCs and fibroblasts derived from these children demonstrated a reduced type I interferon response to HSV-1 challenge and a concomitant enhancement in viral replication [8].
2.2. TLR Expression and Activity
The inflammatory response evoked by viral PAMPs depends on a variety of factors. Firstly, cellular expression of TLRs differs between innate cell types. Human macrophages are known to express high levels of TLR2 and TLR4 while plasmacytoid dendritic cells (pDCs) mainly express TLR7 and TLR9 [1].
Expression patterns also vary between species, where TLR9 is restricted to a few cell types in humans it is widely distributed in mice. Furthermore, expression of certain downstream signaling molecules fluctuates between innate cell types.
For example, pDCs are unique in that they constitutively express the transcription factor IRF7 allowing them to quickly produce high levels of type I IFNs in response to viral infection while other cell types such as macrophages may respond in a more delayed manner [2,3].
Thus, the response to identical viral PAMPs may differ between cell types both in the nature of effector molecules produced and the kinetics of the response. Virally encoded proteins that subvert or distort the TLR response often further complicate this picture. In the subsequent sections we discuss the TLRs individually, detailing the viruses they detect and wherever possible the specific viral products sensed.
2.3. TLR4
The TLR4-mediated response to LPS is well known for its critical role in innate immune control of Gram-negative bacterial infection. It was also the first TLR shown to respond to a viral pathogen. In 2000, Kurt-Jones et al. reported the interaction between the fusion (F) protein of respiratory syncytial virus (RSV) and TLR4 [4]. The importance of TLR4 in human viral disease and RSV pathogenesis has been documented in genetic studies. In humans, inheritances of two different single nucleotide polymorphisms (SNPs) in the ectodomain of TLR4 are associated with reduced responses to both LPS and RSV F. A highly significant association was found between RSV infection in high-risk infants and inheritance of hyporesponsive TLR4 SNPs [5].
This was confirmed in a separate study that likewise found a significant association between these same TLR4 SNPs and severity of RSV disease in infants [6]. Initial studies linking TLR4 expression to RSV pathogenesis were done in the TLR4-deficient mouse strain C57BL10ScNCr (which has a deletion of the gene region containing TLR4) as well as in C3H/HeJ mice (non-signaling point mutation of TLR4) [4,7]. These studies found that RSV activated NF- κB in a TLR4-dependent manner at early time points of infection [8].
The original RSV infection studies with ScNCr mice were controversial as it was suggested that the failure to control RSV was due to a defect in IL-12R signaling [9]. However, this discrepancy between the different studies was due in part to confusion about the mouse nomenclature since the ScNCr mice used in the initial studies (but misidentified as ScCR in the paper [4]) have normal IL-12R [10] while the ScCr mice used by the second group were IL-12R-deficient [9]. More recent work using targeted TLR4 knockouts on a B6 background (with normal IL-12R) have confirmed the role of TLR4 in controlling RSV replication independent of IL-12R, but interestingly these studies have also revealed an even more important role for TLR2 in limiting RSV replication [11].
The purified F protein of RSV induced IL-6 production in a dose-dependent manner in human peripheral blood mononuclear cells (PBMCs) and wild type mouse macrophages alike. However, this response was lost in TLR4 deficient and TLR4 knockout macrophages [4,11]. Studies by Vogel and colleagues have shown that the ability of TLR4 to be triggered by RSV F is critical to prevent RSV-induced pathology. Indeed, the formalin-inactivated RSV vaccine which caused exacerbated disease in clinical trials and was found to contain a denatured, non-stimulatory F protein. The disease enhancing activity of the formalin-inactivated RSV vaccine could be reversed by the addition of MPL, a non-toxic lipid A TLR4 agonist [12]. Disease severity is also correlated with the absence of “alternatively activated” (AA) macrophages that play a crucial role in tissue repair [13].
Taken together with the human and mouse genetics, these studies suggest that TLR4-F protein interactions may protect the host from severe RSV disease by mitigating or reprogramming the host response to promote AA-macrophages and thus promote healing [14]. TLR4 is also important for infections by the retrovirus mouse mammary tumor virus (MMTV).
MMTV was shown to activate NF- κB and induce B220 and CD69 lymphocyte activation markers in B cells from wild type but not C3H/HeJ or congenic BALB/c (C.C3H Tlr4lps-d ) lines [15]. TLR4 activation, attributed to the envelope (Env) protein, was found to stimulate production of IL-10 [16]. Surprisingly induction of TLR4 signaling appears to benefit MMTV. First, it activates quiescent B cells encouraging cell division, which is necessary for viral genome integration in the host chromosome. Secondly, it promotes secretion of IL-10, an immunosuppressive cytokine that helps the virus persist indefinitely [15].
2.4. TLR2
Functional TLR2 exists as a heterodimer with either TLR1 or TLR6 on the plasma membrane of both innate and adaptive immune cells.
It can be activated by lipoteichoic acid, a common component of gram-positive bacteria, as well as GPI anchors of parasitic protozoan such as Plasmodium falciparum. The TLR2/TLR6 heterodimer has recently been shown to play a role in the innate immune response to RSV. Macrophages from mice deficient in TLR2 or TLR6 responded to RSV with lower levels of TNF α , IL-6, CCL2 (MCP-1) and CCL5 (RANTES) than their wild type counterparts. When TLR2 or TLR6 knockout mice were challenged intranasally with RSV they had elevated peak viral titers and lower numbers of neutrophils and activated DC in their lungs [11]. Thus, TLR2/TLR6 signaling likely contributes to both innate immune cell recruitment and viral clearance in vivo during RSV infection [11]. In human PBMCs, TLR2 contributes to IL-8 and MCP-1 production in response to Epstein-Barr virus (EBV) [17]. A TLR2/TLR1-mediated proinflammatory response to the related human cytomegalovirus (HCMV) has also been reported. One study found TLR2 deficient mouse macrophages had significantly reduced IL-6 and IL-8 production in response to UV-inactivated HCMV [18]. Furthermore, expression of TLR2 and CD14 was required for maximal NF- κB activation and IL-8 secretion in HEK293 cells exposed to HCMV. Envelope glycoproteins B and H were later shown to coimmunoprecipitate with TLR2 and TLR1 and are theorized to be the HCMV PAMPs stimulating TLR2 [19]. Lymphocytic Choriomeningitis (LCMV) is a non-cytolytic virus that can cause fatal encephalitis in mice. Wild type glial cells infected with LCMV produce TNF, CCL2 and CCL5, a response that is abolished in cells derived from TLR2 deficient mice [20]. TLR2 also induces MHC class-I and class-II, CD40 and CD86 expression in microglia challenged with LCMV, implicating this pathway in the induction of adaptive immunity [20]. In LCMV infection, where much of the CNS damage is caused by the immune response itself, it remains to be determined if TLR2 signaling is protective or pathological. Interestingly, TLR2 is important for type I IFN induction during LCMV infection but the mechanism is unclear [21]. Although TLR2 is normally not associated with type I IFN induction, a recent study from Barton and colleagues demonstrated that on inflammatory monocytes, TLR2 regulates induction of type I interferon in response to viral but not bacterial ligands [22]. Surprisingly, it appears TLR2 can play either a protective or detrimental role in disease caused by herpes simplex virus (HSV) depending on the context of the infection. Studies using an intraperitoneal infection model found TLR2 deficient neonates were protected from lethal HSV-1 encephalitis compared to wild type mice [23]. Despite having similar viral loads, the TLR2 knockouts demonstrated improved survival, attenuated symptoms and reduced CNS inflammatory lesions. In contrast, TLR2 was shown to work synergistically with TLR9 to promote survival in an intranasal HSV-1 infection model [24]. In addition, TLR2 has been shown to be beneficial in both intraperitoneal and intravaginal HSV-2 infection models [25]. TLR2‟s role in murine HSV infection models may be influenced by factors such as the size of the viral inoculum, the route of administration and the age of the subject. HSV induced two distinct responses; a TLR2-dependent inflammatory cytokine response and a TLR9 and/or non-TLR-dependent type I IFN response. A strong IFN response is necessary to control early virus replication (IFN-deficient mice quickly succumb to infection) and prevent spread from the genital tract to the brain [25]. Once in the brain, however, inflammation is linked to increased mortality [23]. Measles virus (MV) is another infection in which TLR2 signaling may have both favorable and unfavorable effects. Challenging mice with live or UV-inactivated wild type MV induces IL-6 production and CD150 surface expression in mouse macrophages; a response that is impaired in TLR2-deficient cells [26]. Intriguingly, CD150 is required for entry of wild type MV into monocytes, thus immune activation through TLR2 may in fact benefit the virus by conferring susceptibility. This study identified MV hemaglutinin (HA) protein as the viral PAMP triggering TLR2 activation [26]. MV vaccine strains carrying a single asparagine to tyrosine substitution in the HA protein lacked the ability to activate TLR2.
2.5. TLR3
With the exception of neutrophils and pDCs, TLR3 is widely expressed in innate immune cells where it is localized to the endosomal compartment [27,28].
In 2001, Alexopoulou et al. demonstrated that activation of TLR3 signaling by the double stranded RNA analog poly(I:C) contributed to the production of type I IFN and cytokines in macrophages.
Moreover, genomic dsRNA isolated from reovirus was found to activate wild type but not TLR3 deficient splenocytes. The idea that TLR3 could respond to dsRNA, a common viral PAMP, led to intense speculation about its role in the host response to numerous infections. Counterintuitively, a later study found no difference in the survival, viral titers or pathology of TLR3 deficient mice following reovirus challenge [29]. The authors suggested that during in vivo infection, TLR3 may not encounter reovirus dsRNA or that levels may be too low to efficiently activate TLR3 [29]. This study also reported indistinguishable immune responses to LCMV, VSV and MCMV infection in TLR3 deficient and wild type mice [29]. However, other evidence exists suggesting that TLR3 does in fact play a role in controlling MCMV as some studies observed blunted type I IFN and IL-12 production accompanied by higher viral loads in the spleens of mice lacking TLR3 [30,31]. Despite this, only TLR9 deficient mice had significantly decreased survival compared to wild type suggesting TLR9 is more crucial than TLR3 in MCMV infections [30].
A recent study also implicates TLR3 in immune suppression of the related herpes virus HSV-1. Patients with TLR3 dominant negative mutations were found to be more susceptible to herpes simplex encephalitis, a rare but devastating manifestation of HSV-1 infection [32]. The presumed ligand for TLR3 in infections with DNA viruses is dsRNA generated during bidirectional transcription of opposing DNA strands. TLR3 signaling also reduces lethality of encephalomyocarditis virus (EMCV), a ssRNA virus that directly damages heart tissue [33]. TLR3 deficient mice challenged with EMCV had decreased levels of TNF α , IL-6 and IL-1β mRNA in cardiac tissue and a corresponding reduction in inflammatory infiltrate at 3 days post infection [33]. Without TLR3 signaling, EMCV replicated to higher levels in the heart resulting in more rapid and extensive mortality in knockouts [33]. Although this study indicates that the TLR3-mediated inflammatory response is beneficial in EMCV infections; TLR3 signaling appears to be detrimental in a number of other viral infections. For instance, TLR3 deficient mice were protected compared to their wild type counterparts when challenged with a lethal dose of West Nile Virus (WNV) [34]. This study found that TLR3 driven production of inflammatory cytokines compromised the blood-brain barrier facilitating WNV entry. This resulted in higher viral loads in the CNS and worsened neuropathology. Likewise, TLR3 was shown to play a pathologic role in infections with Punta Toro Virus (PTV) [35]. Wild type mice had drastically reduced survival and increased hepatic injury compared to TLR3 deficient mice following PTV challenge. Despite having similar serum and hepatic viral loads, wild type mice had elevated levels of IL-6, IFN γ , CCL2 and CCL5, suggesting these proinflammatory molecules may mediate much of the damage observed [35]. Interestingly, although TLR3 signaling increases inflammation and reduces Influenza A virus (IAV) lung titers, it causes a paradoxical decrease in survival. Thus, in IAV infections, lethality appears to be more dependent on TLR3 signaling than direct virus-induced injury.
2.6. TLR7 and TLR8
TLR7 and TLR8 are two closely related receptors that, like TLR3, act in the endosome.
Human TLR7 and TLR8 were first shown to respond to the imidazoquinoline-like compound resiquimod (R-848), a synthetic drug recognized for its antiviral and antitumor activity [36,37].
We now know that nearly any long single-stranded RNA (ssRNA) is capable of activating TLR7 and TLR8 [38]. Despite this, differences do exist between these receptors. For example, short dsRNAs containing certain motifs preferentially activate TLR7 [39,40]. Furthermore, synthetic agonists specific to TLR7 or TLR8 differentially activate innate immune cells leading to distinct cytokine profiles [41]. In 2004, Diebold et al. showed that TLR7 mediates IFN α production by pDCs in response to live or heat-inactivated influenza virus [42].
This TLR7 response could be elicited simply by exposure to purified genomic ssRNA and was completely abrogated by chloroquine, an inhibitor of endolysosomal acidification [42]. Thus, the authors proposed a model, now known as the exogenous pathway, whereby pDCs endocytose and degrade a portion of incoming influenza virions, allowing TLR7 to engage exposed genomic RNA. A similar TLR7-dependent type I interferon response was observed when pDCs were challenged with vesicular stomatitis virus (VSV) [43]. Under normal circumstances both influenza and VSV require endocytosis for viral entry. However, using a recombinant strain of VSV (VSV-RSV-F), capable of fusing to the plasma membrane, Lund et al. demonstrated that VSV activated TLR7 regardless of the route of viral entry. TLR7 is also responsible for pDC production of IFN α in response to Sendai virus (SV); another ssRNA virus which enters at the plasma membrane [44]. Interestingly studies of SV using human U937 and murine RAW 264.7 myeloid lines found only a partial role for TLR signaling in cytokine and chemokine production [45]. Recent evidence suggests the cytosolic RLR receptors are chiefly responsible for the cytokine and interferon response to SV in myeloid cell types other than pDCs [46]. One important observation gleaned from studies using SV and VSV was that, in contrast to influenza, UV-inactivation of these virions abolished TLR7 activation [44]. From this work a second model of TLR7 activation known as the endogenous pathway was proposed. According to this theory, ssRNA intermediates produced during SV and VSV infection are transferred from the cytoplasm to the endosome by means of autophagy [44]. Thus, to elicit a TLR7 response by this route, cells must be exposed to live, replication competent virus. This model is supported by studies showing that selective inhibitors of autophagy and mice deficient in autophagic pathways lack a TLR7 mediated response to SV and VSV [44]. Recent studies have implicated TLR7 and TLR8 in the response to human immunodeficiency virus (HIV). ssRNA derived from the HIV genome caused murine pDCs and macrophages and human PBMCs to produce IFN α , IL-6 and TNF α [47]. In mice this activity was TLR7-dependent while in humans it appears to rely on TLR8 suggesting that HIV receptors may be species-specific. A study by Wang et al. found IFN α production by human and mouse pDCs responding to Coxsackievirus B (CVB) was also dependent on TLR7 [48]. Interestingly, this response required the presence of CVB-specific antibodies as well as functional Fc Receptor complexes on the pDC surface. Thus they proposed a mechanism whereby opsonized CBV is delivered to the endosome via FcR and once internalized viral RNA is detected by TLR7 [48]. This observation suggests previous exposure to CVB can influence subsequent innate responses furthering our understanding of the complex interplay between adaptive and innate immunity.
2.7. TLR9
In both humans and mice, TLR9 is highly expressed in pDCs, innate cells renowned for their ability to rapidly produce large amounts of type I interferon [1].
TLR9 responds to the unmethylated deoxycytidylate-phosphate-deoxyguanylate (CpG) motifs in viral and bacterial DNA [49]. Not surprisingly TLR9 has been shown to play a crucial role in infections caused by a number of DNA viruses. For instance, TLR9 deficient mice infected with MCMV have a drastically increased mortality compared to their wild type counterparts. This hypersensitivity is likely due to the blunted type I IFN and IL-12 response and reduced NK cell activation which results in an elevated MCMV load [30]. In EBV infection, production of type I IFN, IL-6 and IL-8 by pDCs is largely dependent on TLR9 [17]. This is in contrast to monocytes where TLR2 synergizes with TLR9 to orchestrate the cytokine response to EBV [17]. TLR9 signaling also plays a role in the interferon response to HSV types I and II. One study found IFN α production by mouse pDCs in response to HSV-2 was completely dependent on TLR9 and independent of viral replication [50]. Using cholorquine it was shown that this recognition required endosomal maturation and could be evoked simply by exposure to purified HSV-2 DNA [50]. Furthermore, following in vivo HSV-2 challenge, IFN α was only detectable in the serum of mice with intact TLR9. A similar role for TLR9 was described in the response to HSV-1 by splenic pDCs. However, this study also described a delayed IFN α response by conventional dendritic cells (cDCs) and macrophages that was both TLR9 and MyD88-independent but required exposure to replication competent virus. The TLR9-independent IFN response is likely due to cytoplasmic RLRs and may explain why one study using TLR9 deficient mice identified no in vivo defects in the HSV-1 control [51]. Alternatively, TLR9 signaling may be more important in certain manifestations of HSV-1 induced disease. A recent study showed TLR9 deficient mice did have higher rates of mortality and viral replication when challenged intranasally with HSV-1 [24]. Thus TLR9‟s precise role in HSV pathogenesis and the relative contributions of other PRRs requires further investigation. Figure 1 illustrates the TLRs activated by viral pathogens and depicts their downstream signal pathways.
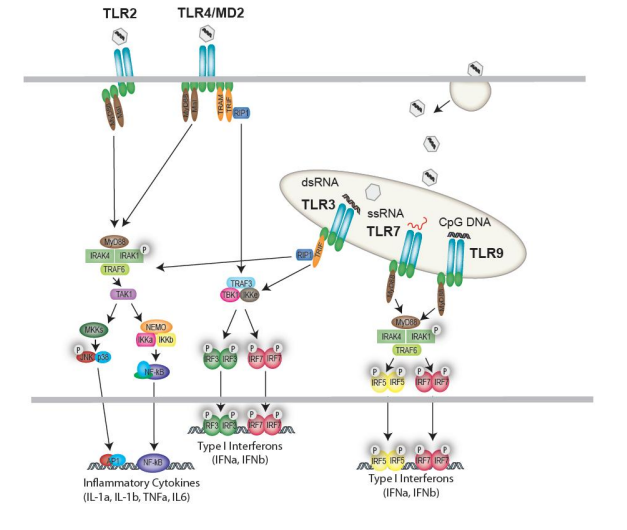
Figure 1. Cell surface and endosomal recognition of viruses by Toll-like receptors (TLRs). TLR2 responds to a variety of viruses resulting in activation of a MyD88-dependent NF-κB and MAPK pathway. TLR4, responding to viral proteins (e.g., RSV F-protein) activates both a MyD88-dependent and MyD88-independent response. The MyD88- dependent response leads to transcriptional regulation of inflammatory cytokines, while the MyD88-independent response is regulated via TRAM/TRIF and the IKK-related kinases which drive IRF3 activation and type I Interferon production. In the endosome, TLR3, TLR7, TLR8 and TLR9 sense viral nucleic acids and generate either IRF3 activation (TLR3) or IRF7-driven type I IFNs (TLR7, 8 and 9).